

Mutation of Proteolipid Protein 1 Gene: From Severe Hypomyelinating Leukodystrophy to Inherited Spastic Paraplegia
- PMID: 35885014
- PMCID: PMC9313024
- DOI: 10.3390/biomedicines10071709
Mutation of Proteolipid Protein 1 Gene: From Severe Hypomyelinating Leukodystrophy to Inherited Spastic Paraplegia
Abstract
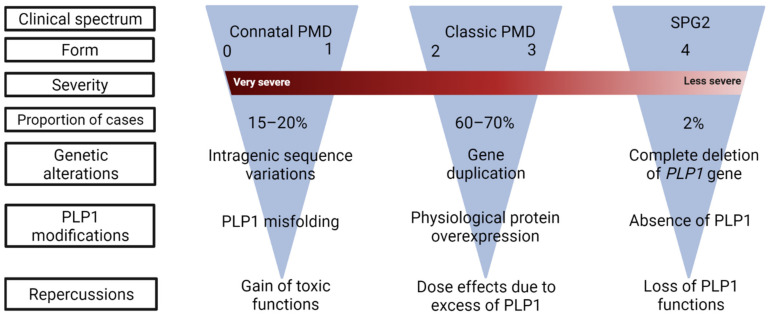
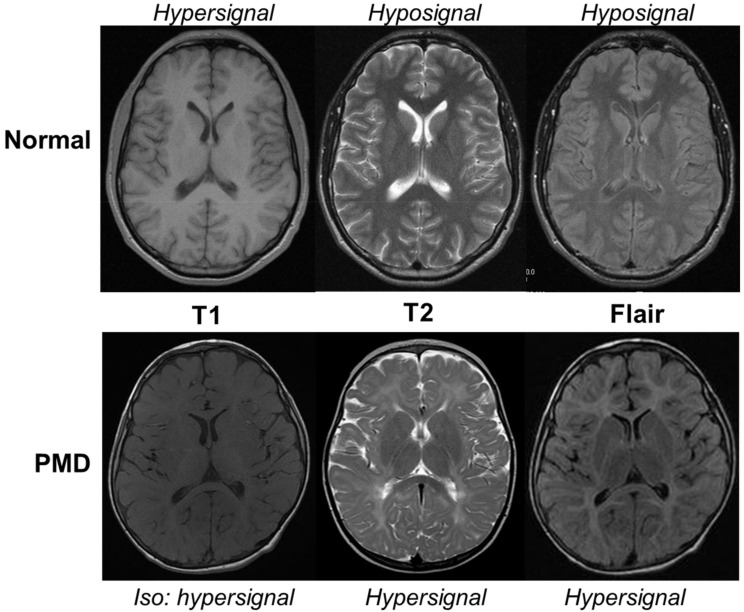
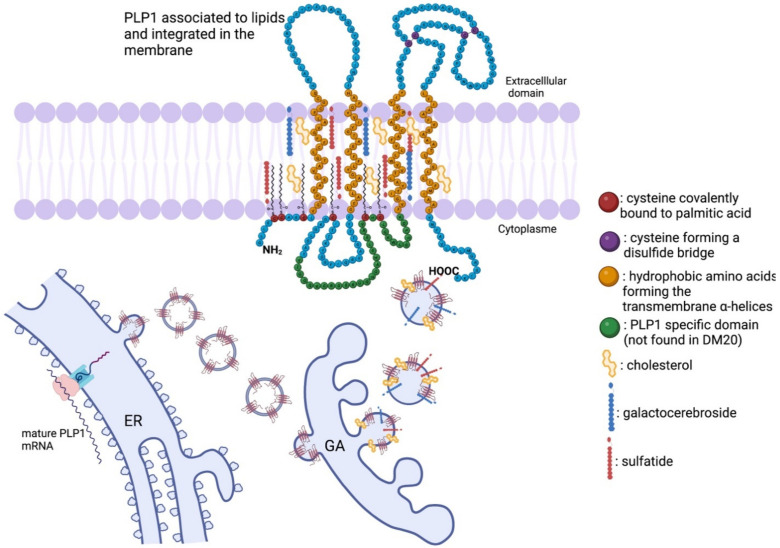
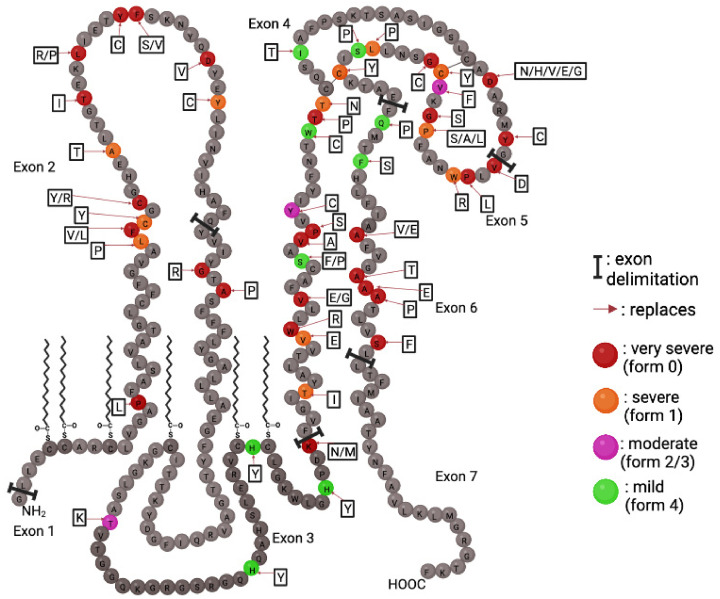
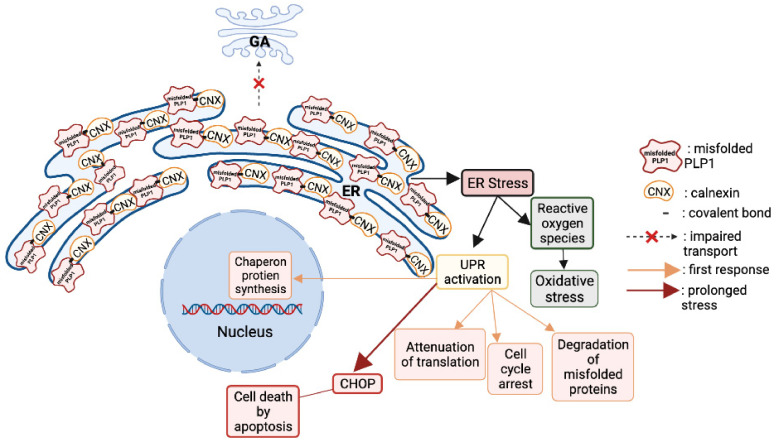
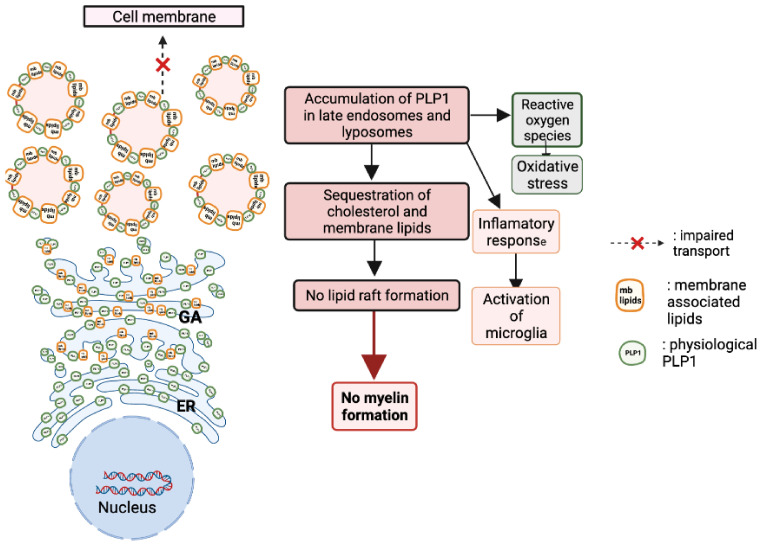
Pelizaeus-Merzbacher Disease (PMD) is an inherited leukodystrophy affecting the central nervous system (CNS)-a rare disorder that especially concerns males. Its estimated prevalence is 1.45-1.9 per 100,000 individuals in the general population. Patients affected by PMD exhibit a drastic reduction or absence of myelin sheaths in the white matter areas of the CNS. The Proteolipid Protein 1 (PLP1) gene encodes a transmembrane proteolipid protein. PLP1 is the major protein of myelin, and it plays a key role in the compaction, stabilization, and maintenance of myelin sheaths. Its function is predominant in oligodendrocyte development and axonal survival. Mutations in the PLP1 gene cause the development of a wide continuum spectrum of leukopathies from the most severe form of PMD for whom patients exhibit severe CNS hypomyelination to the relatively mild late-onset type 2 spastic paraplegia, leading to the concept of PLP1-related disorders. The genetic diversity and the biochemical complexity, along with other aspects of PMD, are discussed to reveal the obstacles that hinder the development of treatments. This review aims to provide a clinical and mechanistic overview of this spectrum of rare diseases.
Keywords: Pelizaeus-Merzbacher disease (PMD); animal models; diagnosis; proteolipid protein 1 variants; spastic paraplegia (SPG2); treatments.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Raine C.S. Myelin. Springer; Berlin/Heidelberg, Germany: 1984. Morphology of myelin and myelination; pp. 1–50.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
