

Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat
- PMID: 35885895
- PMCID: PMC9317296
- DOI: 10.3390/genes13071112
Characterization of Expression and Epigenetic Features of Core Genes in Common Wheat
Abstract
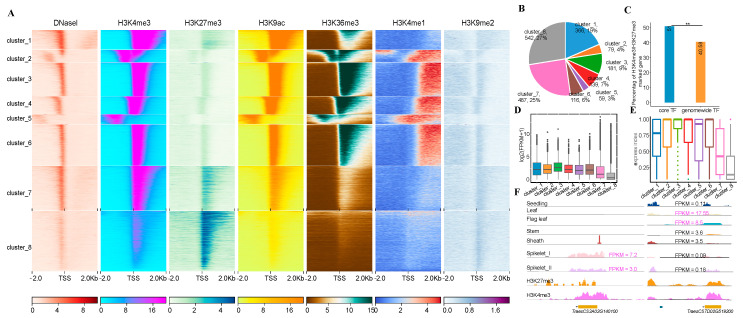
The availability of multiple wheat genome sequences enables us to identify core genes and characterize their genetic and epigenetic features, thereby advancing our understanding of their biological implications within individual plant species. It is, however, largely understudied in wheat. To this end, we reanalyzed genome sequences from 16 different wheat varieties and identified 62,299 core genes. We found that core and non-core genes have different roles in subgenome differentiation. Meanwhile, according to their expression profiles, these core genes can be classified into genes related to tissue development and stress responses, including 3376 genes highly expressed in both spikelets and at high temperatures. After associating with six histone marks and open chromatin, we found that these core genes can be divided into eight sub-clusters with distinct epigenomic features. Furthermore, we found that ca. 51% of the expressed transcription factors (TFs) were marked with both H3K27me3 and H3K4me3, indicative of the bivalency feature, which can be involved in tissue development through the TF-centered regulatory network. Thus, our study provides a valuable resource for the functional characterization of core genes in stress responses and tissue development in wheat.
Keywords: Triticum aestivum; core genes; epigenetic features; expression.
Conflict of interest statement
The authors declare no conflict of interest.
Figures




References
-
- Au K.F., Valiente-Mullor C., Beamud B., Ansari I., Francés-Cuesta C., García-González N., Mejía L., Ruiz-Hueso P., González-Candelas F. One is not enough: On the effects of reference genome for the mapping and subsequent analyses of short-reads. PLoS Comput. Biol. 2021;17:e1008678. doi: 10.1371/journal.pcbi.1008678. - DOI - PMC - PubMed
-
- Tettelin H., Masignani V., Cieslewicz M.J., Donati C., Medini D., Ward N.L., Angiuoli S.V., Crabtree J., Jones A.L., Durkin A.S., et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA. 2005;102:13950–13955. doi: 10.1073/pnas.0506758102. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
