

Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization
- PMID: 35889075
- PMCID: PMC9320768
- DOI: 10.3390/microorganisms10071356
Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization
Abstract
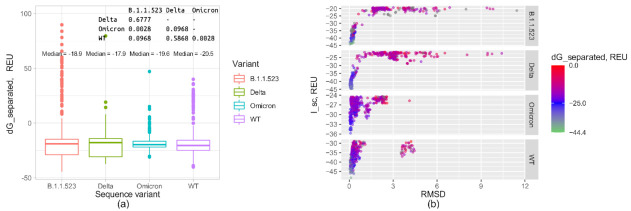
Here, we report the emergence of the variant lineage B.1.1.523 that contains a set of mutations including 156_158del, E484K and S494P in the spike protein. E484K and S494P are known to significantly reduce SARS-CoV-2 neutralization by convalescent and vaccinated sera and are considered as mutations of concern. Lineage B.1.1.523 presumably originated in the Russian Federation and spread across European countries with the peak of transmission in April-May 2021. The B.1.1.523 lineage has now been reported from 31 countries. In this article, we analyze the possible origin of this mutation subset and its immune response using in silico methods.
Keywords: B.1.1.523; GISAID; SARS-CoV-2; phylogeny; variant of concern (VOC).
Conflict of interest statement
The authors declare no conflict of interest.
Figures











References
-
- World Health Organisation WHO Coronavirus (COVID-19) Dashboard. [(accessed on 1 October 2021)]. Available online: https://covid19.who.int/
-
- European Centre for Disease Prevention and Control How ECDC Collects and Processes COVID-19 Data. [(accessed on 1 October 2021)]. Available online: https://www.ecdc.europa.eu/en/covid-19/data-collection.
-
- World Health Organisation Tracking SARS-CoV-2 Variants. [(accessed on 1 October 2021)]. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/
LinkOut - more resources
Full Text Sources
Miscellaneous
