

Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease
- PMID: 35892340
- PMCID: PMC9394283
- DOI: 10.3390/biom12081030
Molecular, Subcellular, and Arrhythmogenic Mechanisms in Genetic RyR2 Disease
Abstract
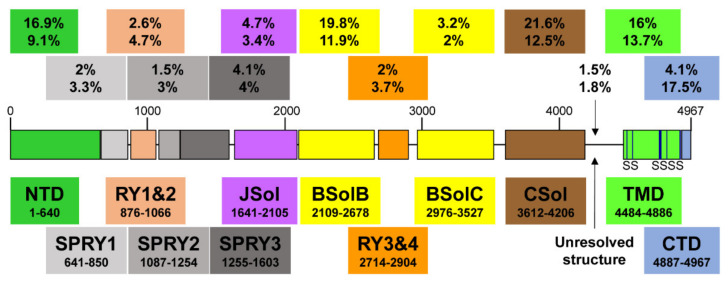
The ryanodine receptor (RyR2) has a critical role in controlling Ca2+ release from the sarcoplasmic reticulum (SR) throughout the cardiac cycle. RyR2 protein has multiple functional domains with specific roles, and four of these RyR2 protomers are required to form the quaternary structure that comprises the functional channel. Numerous mutations in the gene encoding RyR2 protein have been identified and many are linked to a wide spectrum of arrhythmic heart disease. Gain of function mutations (GoF) result in a hyperactive channel that causes excessive spontaneous SR Ca2+ release. This is the predominant cause of the inherited syndrome catecholaminergic polymorphic ventricular tachycardia (CPVT). Recently, rare hypoactive loss of function (LoF) mutations have been identified that produce atypical effects on cardiac Ca2+ handling that has been termed calcium release deficiency syndrome (CRDS). Aberrant Ca2+ release resulting from both GoF and LoF mutations can result in arrhythmias through the Na+/Ca2+ exchange mechanism. This mini-review discusses recent findings regarding the role of RyR2 domains and endogenous regulators that influence RyR2 gating normally and with GoF/LoF mutations. The arrhythmogenic consequences of GoF/LoF mutations will then be discussed at the macromolecular and cellular level.
Keywords: arrhythmias; calcium release deficiency syndrome; calcium sparks; catecholaminergic polymorphic ventricular tachycardia; delayed afterdepolarizations; early afterdepolarizations; long QT syndrome; ryanodine receptor.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
