

CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases
- PMID: 35894054
- PMCID: PMC9330826
- DOI: 10.3390/pathogens11080831
CD4+ Cytotoxic T Cells Involved in the Development of EBV-Associated Diseases
Abstract
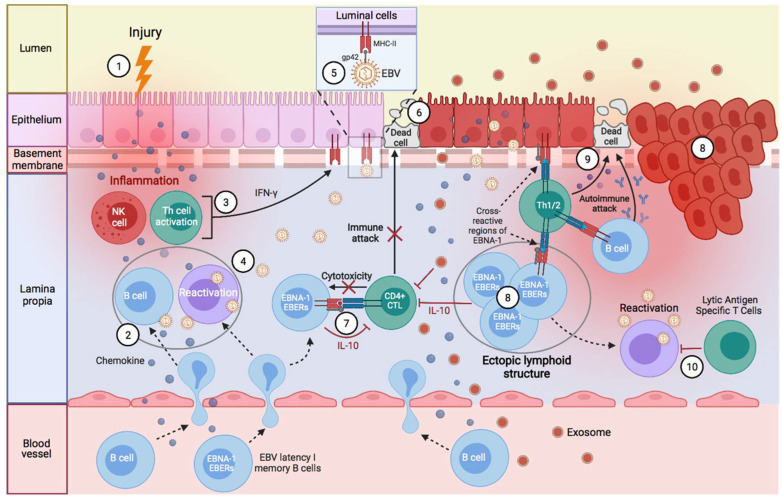
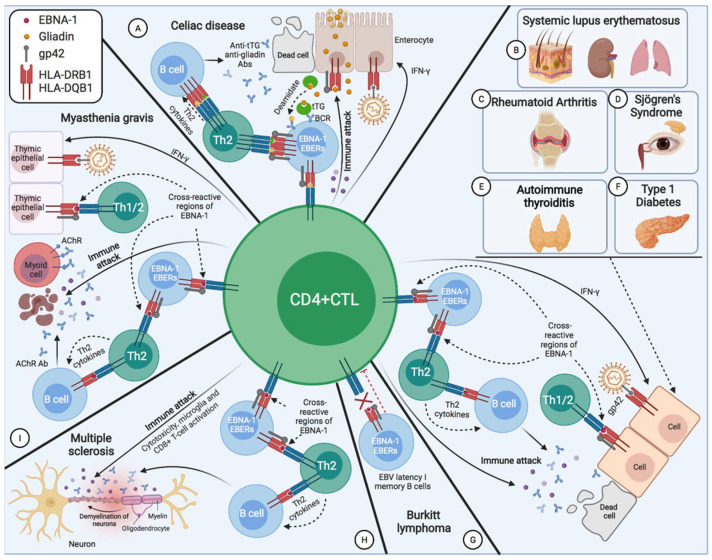
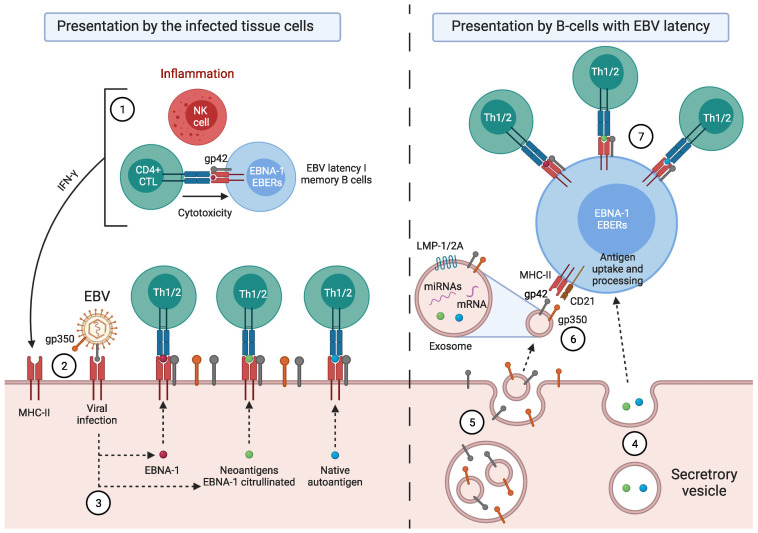
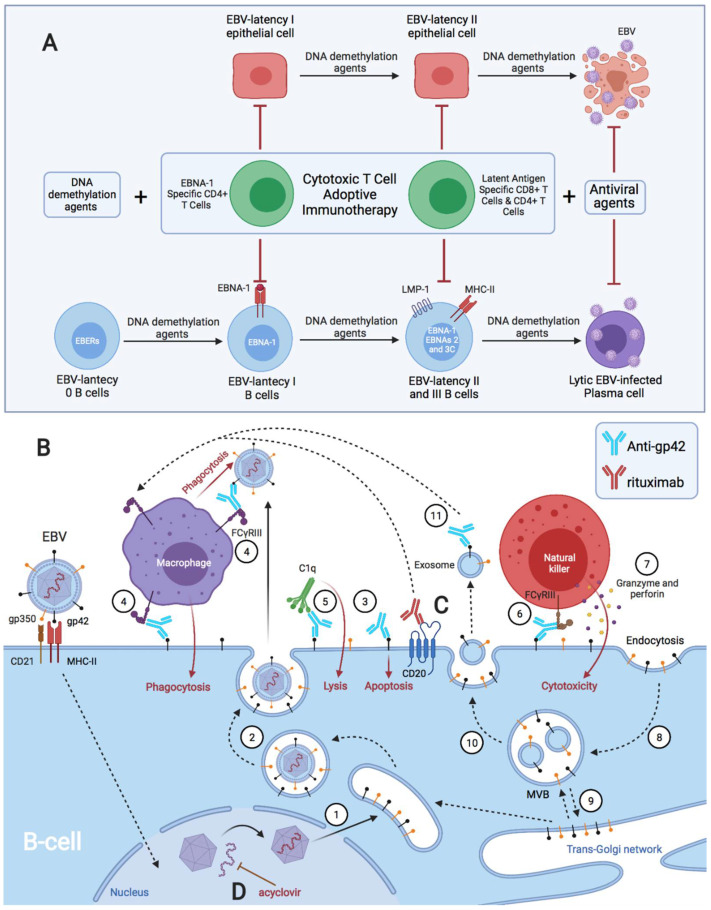
Activated cytotoxic CD4 T cells (HLA-DR+) play an important role in the control of EBV infection, especially in cells with latency I (EBNA-1). One of the evasion mechanisms of these latency cells is generated by gp42, which, via peripherally binding to the β1 domain of the β chain of MHC class II (HLA-DQ, -DR, and -DP) of the infected B lymphocyte, can block/alter the HLA class II/T-cell receptor (TCR) interaction, and confer an increased level of susceptibility towards the development of EBV-associated autoimmune diseases or cancer in genetically predisposed individuals (HLA-DRB1* and DQB1* alleles). The main developments predisposing the factors of these diseases are: EBV infection; HLA class II risk alleles; sex; and tissue that is infiltrated with EBV-latent cells, forming ectopic lymphoid structures. Therefore, there is a need to identify treatments for eliminating cells with EBV latency, because the current treatments (e.g., antivirals and rituximab) are ineffective.
Keywords: CD4+ CTL; DQB1; DRB1; EBV EBNA-1; Gp42; HLA; autoimmunity; cancer.
Conflict of interest statement
The author declares no conflict of interest.
Figures
Similar articles
-
HLA-DQ β1 alleles associated with Epstein-Barr virus (EBV) infectivity and EBV gp42 binding to cells.JCI Insight. 2017 Feb 23;2(4):e85687. doi: 10.1172/jci.insight.85687. JCI Insight. 2017. PMID: 28239644 Free PMC article.
-
Molecular characterization of major histocompatibility complex class II gene expression and demonstration of antigen-specific T cell response indicate a new phenotype in class II-deficient patients.J Exp Med. 1995 Apr 1;181(4):1411-23. doi: 10.1084/jem.181.4.1411. J Exp Med. 1995. PMID: 7699327 Free PMC article.
-
Comprehensive Analysis of CD4+ T Cell Responses to CMV pp65 Antigen Restricted by Single HLA-DR, -DQ, and -DP Allotype Within an Individual.Front Immunol. 2021 Feb 15;11:602014. doi: 10.3389/fimmu.2020.602014. eCollection 2020. Front Immunol. 2021. PMID: 33658991 Free PMC article.
-
HLA-A0201 and HLA-B7 binding peptides in the EBV-encoded EBNA-1, EBNA-2 and BZLF-1 proteins detected in the MHC class I stabilization assay. Low proportion of binding motifs for several HLA class I alleles in EBNA-1.Int Immunol. 1995 Apr;7(4):653-63. doi: 10.1093/intimm/7.4.653. Int Immunol. 1995. PMID: 7547693
-
HLA class II sequences and genetic susceptibility to insulin dependent diabetes mellitus.Baillieres Clin Endocrinol Metab. 1991 Sep;5(3):395-411. doi: 10.1016/s0950-351x(05)80138-7. Baillieres Clin Endocrinol Metab. 1991. PMID: 1909860 Review.
Cited by
-
Epstein-Barr virus-acquired immunodeficiency in myalgic encephalomyelitis-Is it present in long COVID?J Transl Med. 2023 Sep 17;21(1):633. doi: 10.1186/s12967-023-04515-7. J Transl Med. 2023. PMID: 37718435 Free PMC article. Review.
-
Hypocortisolemic ASIA: a vaccine- and chronic infection-induced syndrome behind the origin of long COVID and myalgic encephalomyelitis.Front Immunol. 2024 Jul 9;15:1422940. doi: 10.3389/fimmu.2024.1422940. eCollection 2024. Front Immunol. 2024. PMID: 39044822 Free PMC article. Review.
-
Cytotoxic CD4+ T cells in chronic viral infections and cancer.Front Immunol. 2023 Oct 25;14:1271236. doi: 10.3389/fimmu.2023.1271236. eCollection 2023. Front Immunol. 2023. PMID: 37965314 Free PMC article. Review.
References
-
- Hintzen R.Q., de Jong R., Lens S.M., Brouwer M., Baars P., van Lier R.A. Regulation of CD27 Expression on Subsets of Mature T-Lymphocytes. [(accessed on 8 January 2021)];J. Immunol. 1993 151:2426–2435. Available online: https://pubmed.ncbi.nlm.nih.gov/7689607/ - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
