

Evolutionary Genomics of a Subdivided Species
- PMID: 35895848
- PMCID: PMC9372567
- DOI: 10.1093/molbev/msac152
Evolutionary Genomics of a Subdivided Species
Abstract
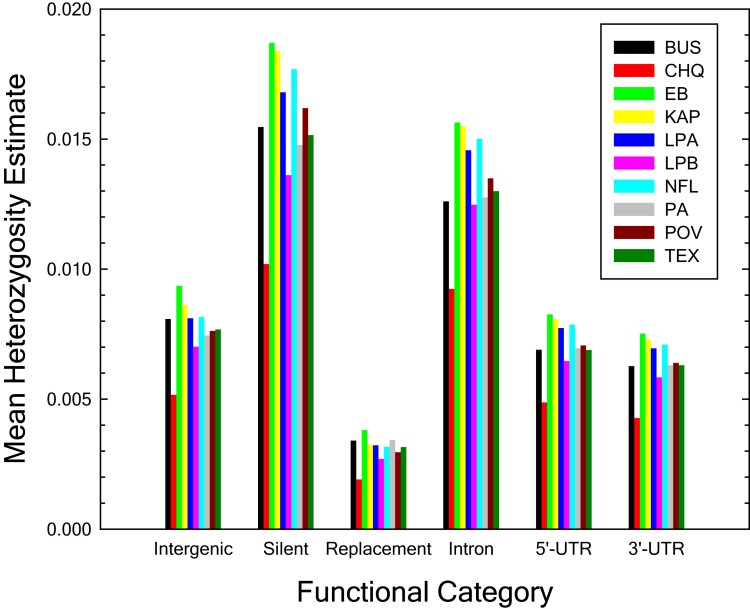
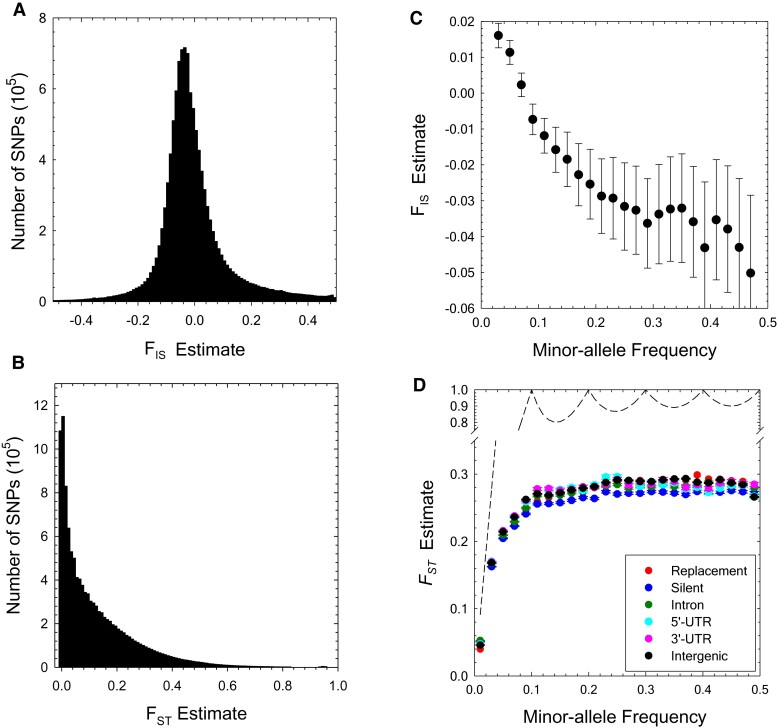
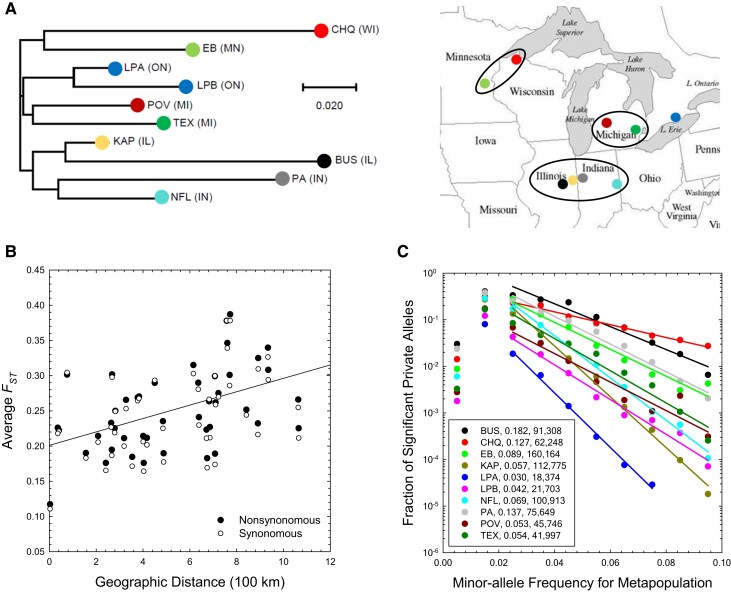
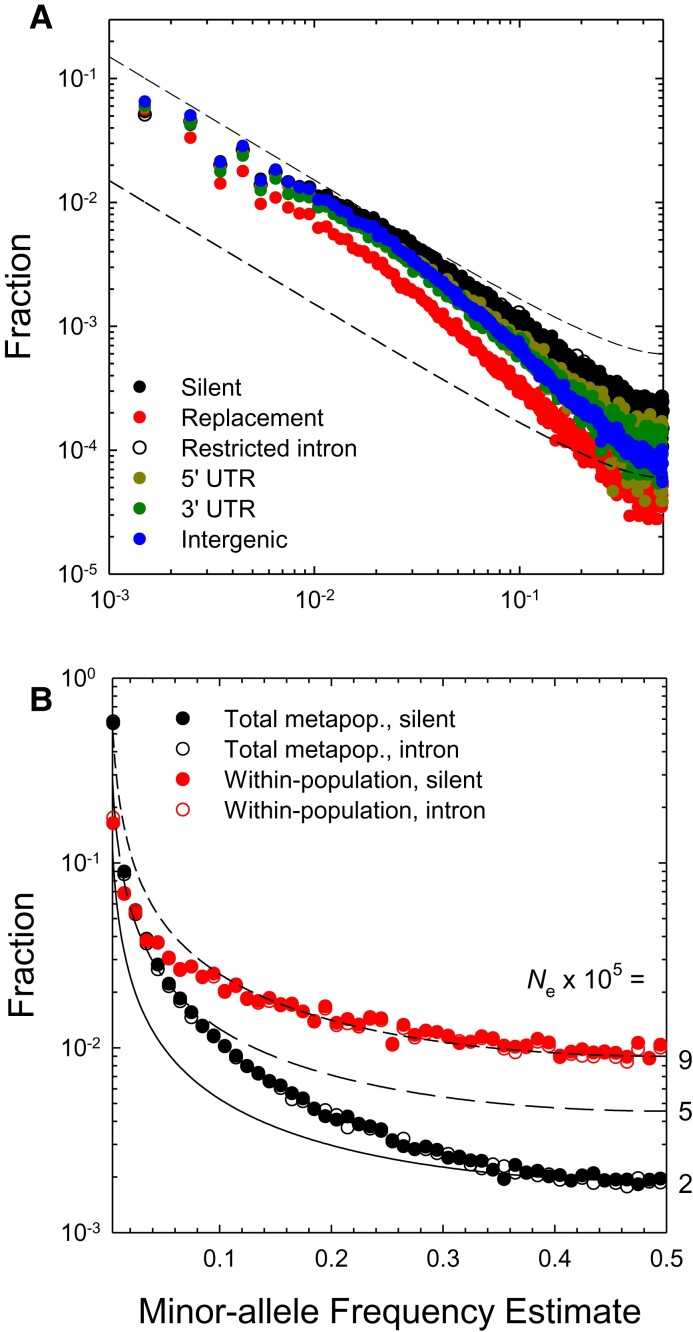
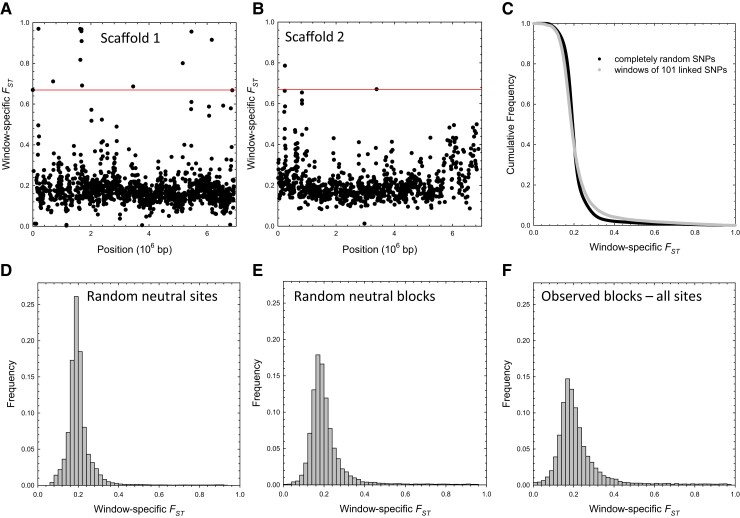
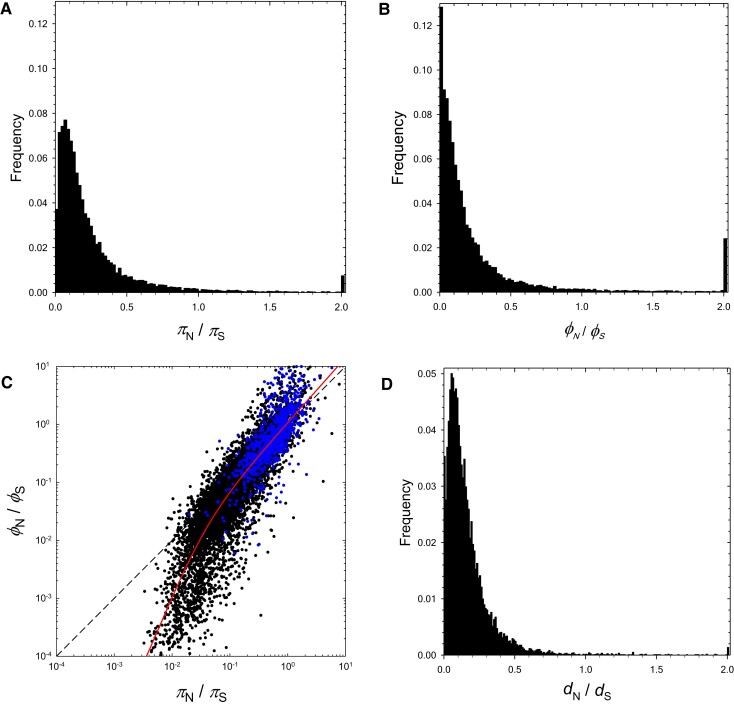
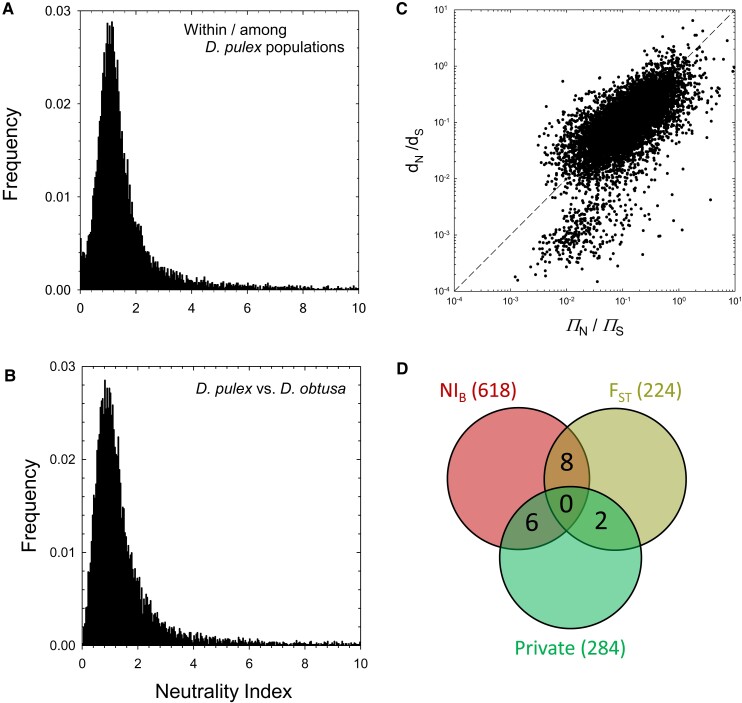
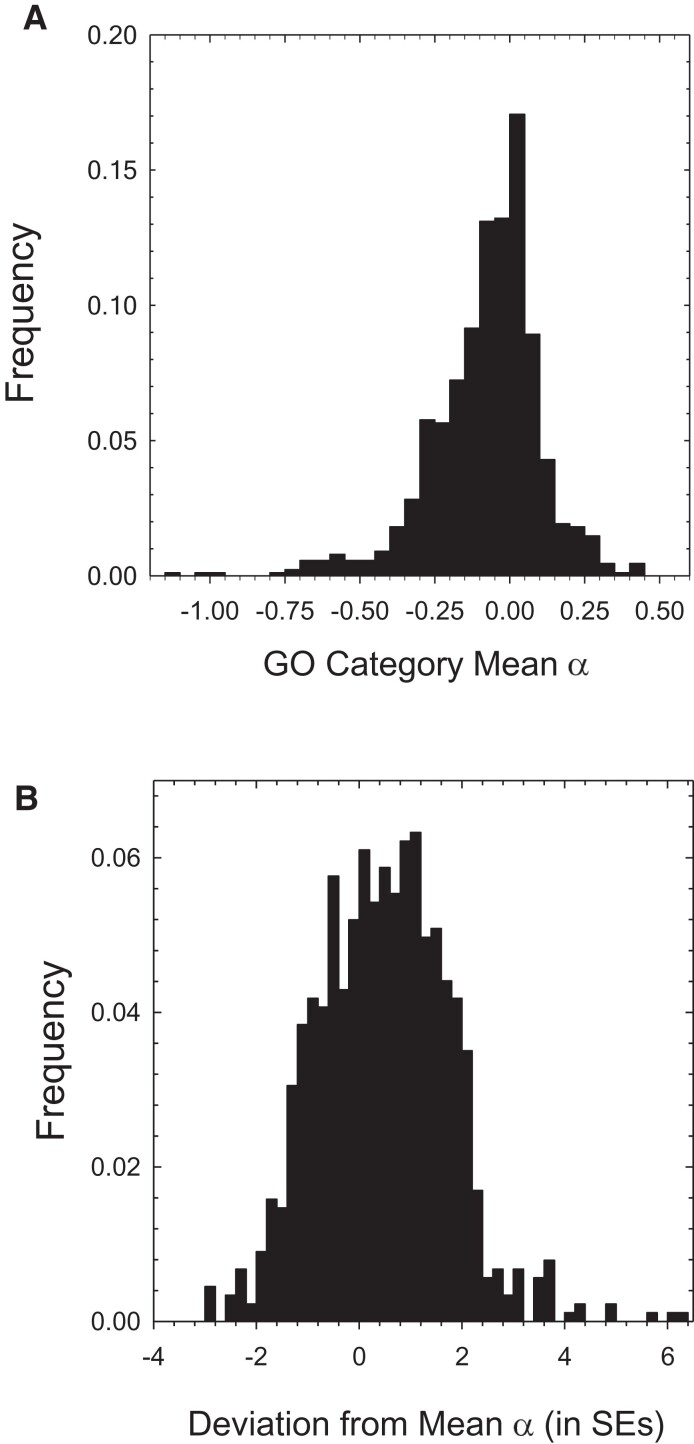
The ways in which genetic variation is distributed within and among populations is a key determinant of the evolutionary features of a species. However, most comprehensive studies of these features have been restricted to studies of subdivision in settings known to have been driven by local adaptation, leaving our understanding of the natural dispersion of allelic variation less than ideal. Here, we present a geographic population-genomic analysis of 10 populations of the freshwater microcrustacean Daphnia pulex, an emerging model system in evolutionary genomics. These populations exhibit a pattern of moderate isolation-by-distance, with an average migration rate of 0.6 individuals per generation, and average effective population sizes of ∼650,000 individuals. Most populations contain numerous private alleles, and genomic scans highlight the presence of islands of excessively high population subdivision for more common alleles. A large fraction of such islands of population divergence likely reflect historical neutral changes, including rare stochastic migration and hybridization events. The data do point to local adaptive divergence, although the precise nature of the relevant variation is diffuse and cannot be associated with particular loci, despite the very large sample sizes involved in this study. In contrast, an analysis of between-species divergence highlights positive selection operating on a large set of genes with functions nearly nonoverlapping with those involved in local adaptation, in particular ribosome structure, mitochondrial bioenergetics, light reception and response, detoxification, and gene regulation. These results set the stage for using D. pulex as a model for understanding the relationship between molecular and cellular evolution in the context of natural environments.
Keywords: Daphnia pulex; local adaptation; neutrality index; population genomics; population structure; population subdivision; private alleles; site-frequency spectrum.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
-
- Agar WE. 1920. The genetics of a Daphnia hybrid during parthenogenesis. J Genet. 10:303–330.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
