

AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer
- PMID: 35895872
- PMCID: PMC9627128
- DOI: 10.1158/2159-8290.CD-22-0111
AXL and Error-Prone DNA Replication Confer Drug Resistance and Offer Strategies to Treat EGFR-Mutant Lung Cancer
Abstract
Anticancer therapies have been limited by the emergence of mutations and other adaptations. In bacteria, antibiotics activate the SOS response, which mobilizes error-prone factors that allow for continuous replication at the cost of mutagenesis. We investigated whether the treatment of lung cancer with EGFR inhibitors (EGFRi) similarly engages hypermutators. In cycling drug-tolerant persister (DTP) cells and in EGFRi-treated patients presenting residual disease, we observed upregulation of GAS6, whereas ablation of GAS6's receptor, AXL, eradicated resistance. Reciprocally, AXL overexpression enhanced DTP survival and accelerated the emergence of T790M, an EGFR mutation typical to resistant cells. Mechanistically, AXL induces low-fidelity DNA polymerases and activates their organizer, RAD18, by promoting neddylation. Metabolomics uncovered another hypermutator, AXL-driven activation of MYC, and increased purine synthesis that is unbalanced by pyrimidines. Aligning anti-AXL combination treatments with the transition from DTPs to resistant cells cured patient-derived xenografts. Hence, similar to bacteria, tumors tolerate therapy by engaging pharmacologically targetable endogenous mutators.
Significance: EGFR-mutant lung cancers treated with kinase inhibitors often evolve resistance due to secondary mutations. We report that in similarity to the bacterial SOS response stimulated by antibiotics, endogenous mutators are activated in drug-treated cells, and this heralds tolerance. Blocking the process prevented resistance in xenograft models, which offers new treatment strategies. This article is highlighted in the In This Issue feature, p. 2483.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures



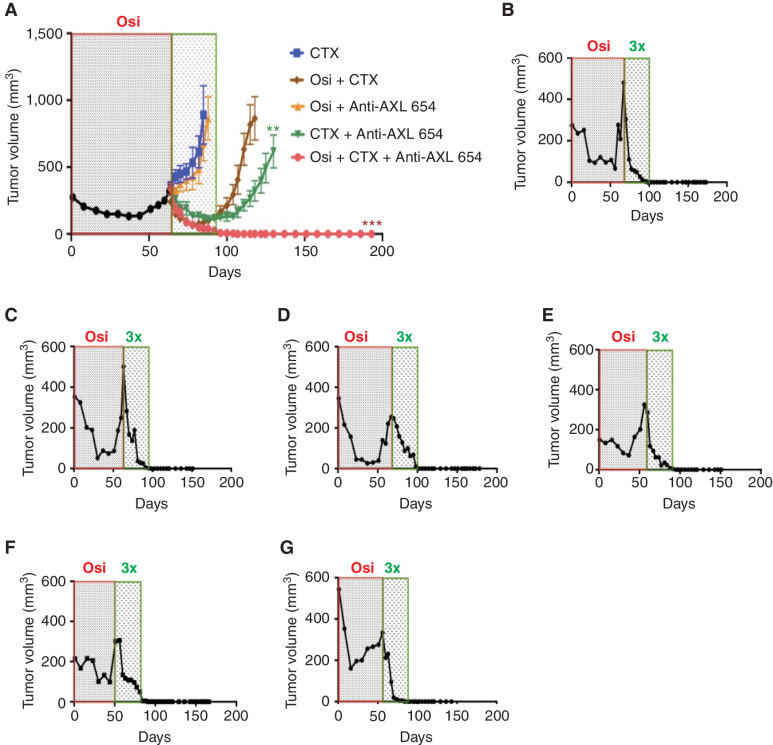
![Figure 4. AXL induces purine metabolism and increases PuMB. A, RNA from PC9 cells and AXL-KO cells was sequenced, and differentially expressed genes were subjected to KEGG Pathway Enrichment analysis. B, A waterfall plot depicting fold changes in metabolite abundance in AXL-KO relative to PC9-WT (control) cells. Metabolites were rank-ordered, and specific compounds are indicated (assays performed in triplicates). C, Fractional labeling of IMP in PC9-KO and PC9-WT (control) cells using [amide-15N]glutamine and 24 hours of incubation. D, Fractional labeling of purines determined in AXL-KO and PC9-WT cells incubated for 24 hours with [U-13C]glucose (means ± SD; 3 experiments). E, An AXL expression vector and MYC promoter reporter plasmid were transfected into HEK293 cells. Renilla was used as a control. Luminescence reading was taken 48 hours later. F, PPAT and PAICS promoter reporter plasmids were transfected into HEK293 cells, along with AXL and MYC vectors. Luminescence readings (means ± SD; 3 experiments) were normalized to a GAPDH reporter. G, PC9 cells were treated with osimertinib (1 μmol/L) or DMSO for the indicated time intervals. RNA was isolated and subjected to real-time PCR using primers corresponding to the indicated transcripts. H, PC9 cells were either untreated or treated for 9 days with TKIs at the indicated concentrations. Control cells were treated with DMSO. The indicated proteins were detected using immunoblotting. Tubulin served as the gel loading control. I, PuMB was analyzed in the TCGA lung adenocarcinoma data set (n = 506 patients) and presented versus EGFR's mutational status. J, A cohort of 10 treatment-naïve patients, for whom both tumoral single-cell RNA-seq data and tumor whole-exome sequencing data were available, was analyzed for AXL expression and PuMB. Dots show individual data points and the diagonal represents a regression line. K, The status of AXL expression level and presence of EGFR or RAS mutations in the TCGA data set are shown. L, A core MYC gene-expression signature was analyzed against the level of AXL expression in the cohort of 506 patients. ***, P ≤ 0.001.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/0057/9627128/578ffdabaaa6/2666fig4.jpg)

Comment in
- doi: 10.1158/2159-8290.CD-12-11-ITI
References
-
- Radman M. SOS repair hypothesis: phenomenology of an inducible DNA repair which is accompanied by mutagenesis. Basic Life Sci 1975;5A:355–67. - PubMed
-
- Baharoglu Z, Mazel D. SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 2014;38:1126–45. - PubMed
-
- Russo M, Crisafulli G, Sogari A, Reilly NM, Arena S, Lamba S, et al. . Adaptive mutability of colorectal cancers in response to targeted therapies. Science 2019;366:1473–80. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
