

Proteomic and phosphoproteomic landscapes of acute myeloid leukemia
- PMID: 35895896
- PMCID: PMC9523374
- DOI: 10.1182/blood.2022016033
Proteomic and phosphoproteomic landscapes of acute myeloid leukemia
Abstract
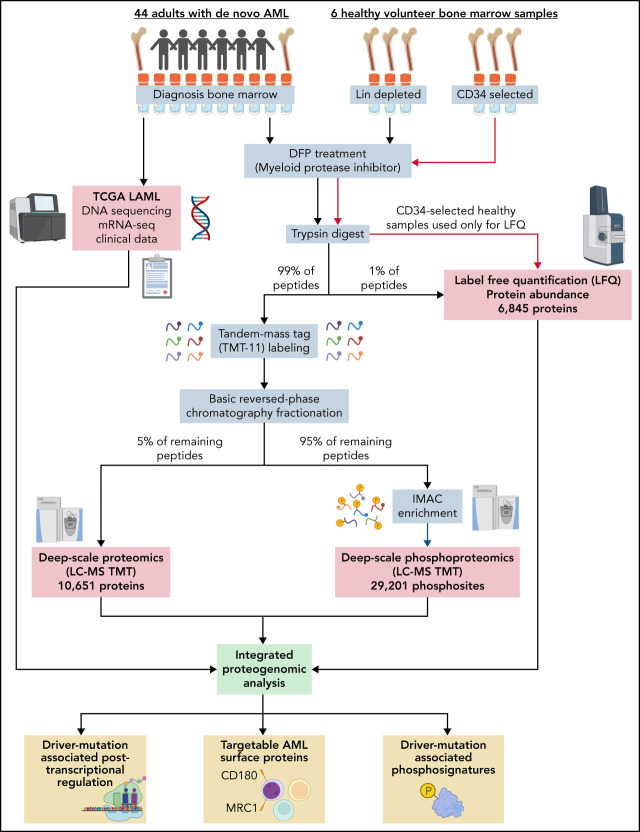
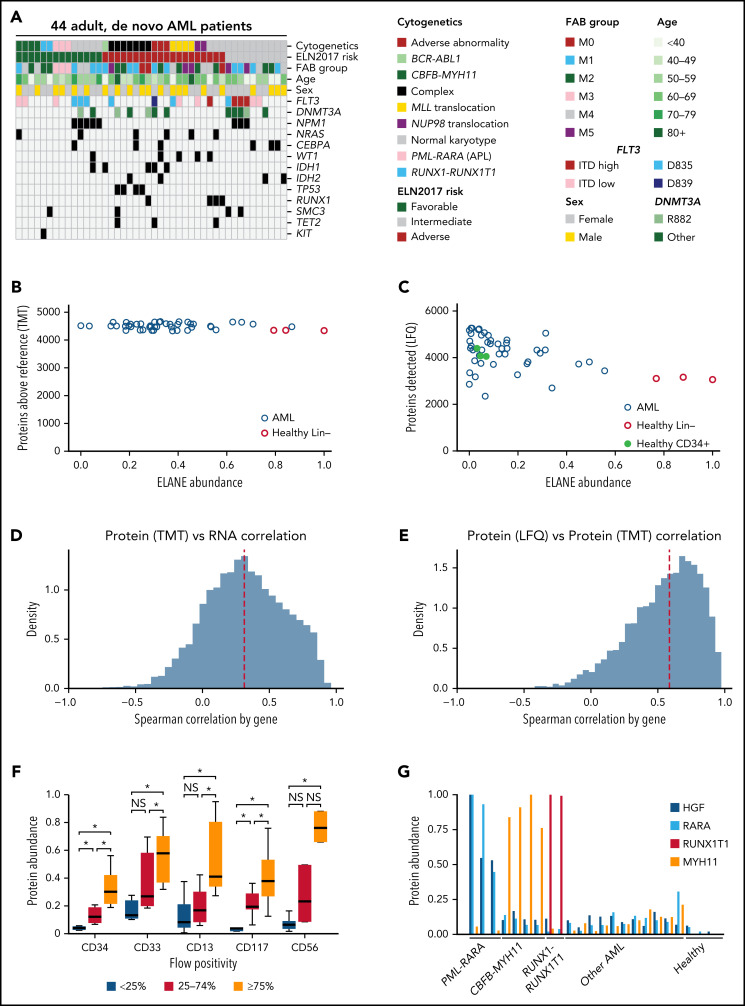
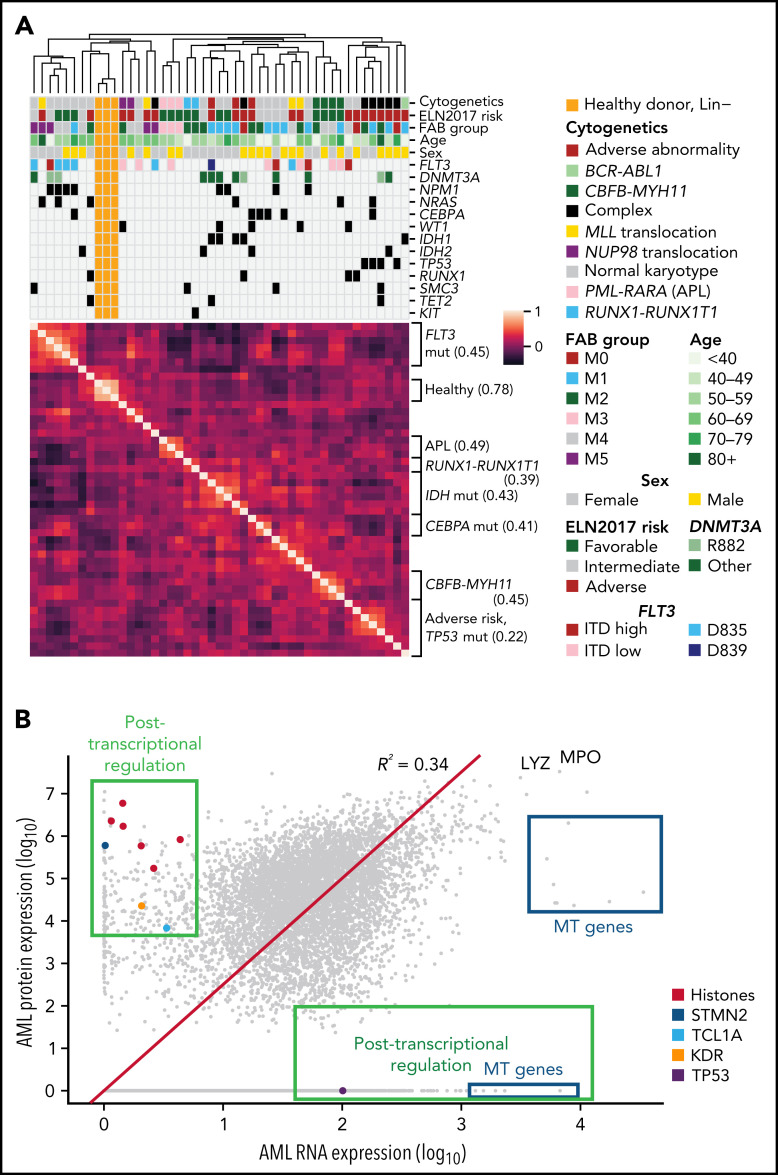
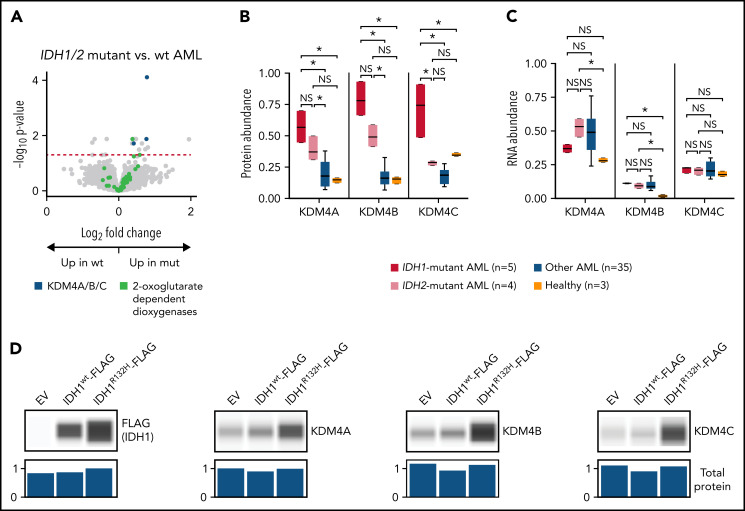
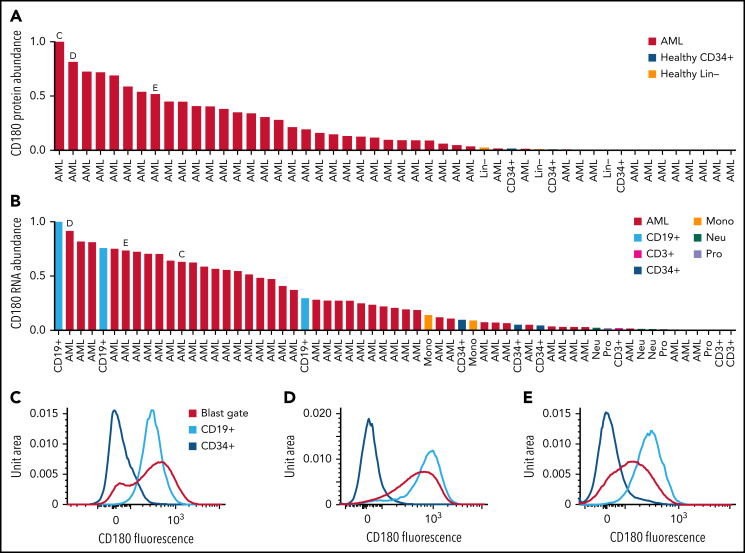
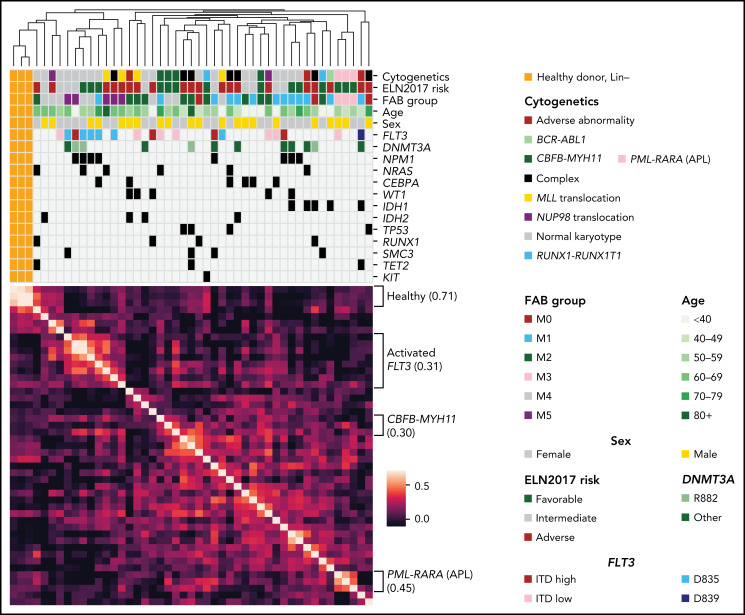
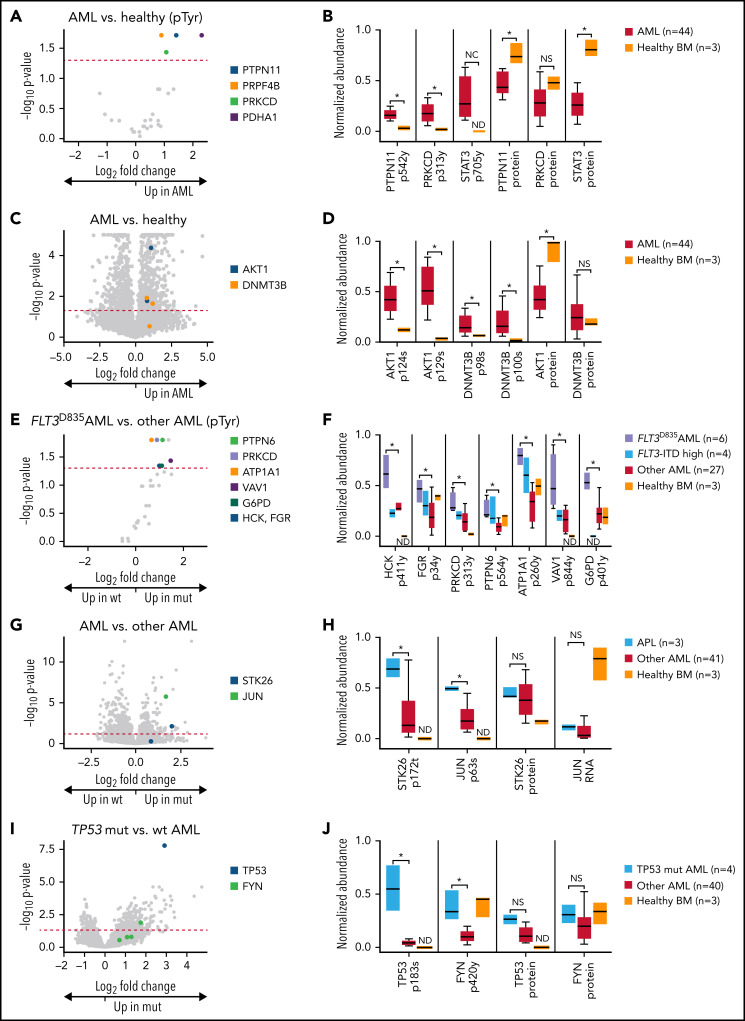
We have developed a deep-scale proteome and phosphoproteome database from 44 representative acute myeloid leukemia (AML) patients from the LAML TCGA dataset and 6 healthy bone marrow-derived controls. After confirming data quality, we orthogonally validated several previously undescribed features of AML revealed by the proteomic data. We identified examples of posttranscriptionally regulated proteins both globally (ie, in all AML samples) and also in patients with recurrent AML driver mutations. For example, samples with IDH1/2 mutations displayed elevated levels of the 2-oxoglutarate-dependent histone demethylases KDM4A/B/C, despite no changes in messenger RNA levels for these genes; we confirmed this finding in vitro. In samples with NPMc mutations, we identified several nuclear importins with posttranscriptionally increased protein abundance and showed that they interact with NPMc but not wild-type NPM1. We identified 2 cell surface proteins (CD180 and MRC1/CD206) expressed on AML blasts of many patients (but not healthy CD34+ stem/progenitor cells) that could represent novel targets for immunologic therapies and confirmed these targets via flow cytometry. Finally, we detected nearly 30 000 phosphosites in these samples; globally, AML samples were associated with the abnormal phosphorylation of specific residues in PTPN11, STAT3, AKT1, and PRKCD. FLT3-TKD samples were associated with increased phosphorylation of activating tyrosines on the cytoplasmic Src-family tyrosine kinases FGR and HCK and related signaling proteins. PML-RARA-initiated AML samples displayed a unique phosphorylation signature, and TP53-mutant samples showed abundant phosphorylation of serine-183 on TP53 itself. This publicly available database will serve as a foundation for further investigations of protein dysregulation in AML pathogenesis.
© 2022 by The American Society of Hematology.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
