

Current View on the Molecular Mechanisms Underlying Fibrin(ogen)-Dependent Inflammation
- PMID: 35896433
- PMCID: PMC10680782
- DOI: 10.1055/a-1910-4538
Current View on the Molecular Mechanisms Underlying Fibrin(ogen)-Dependent Inflammation
Abstract
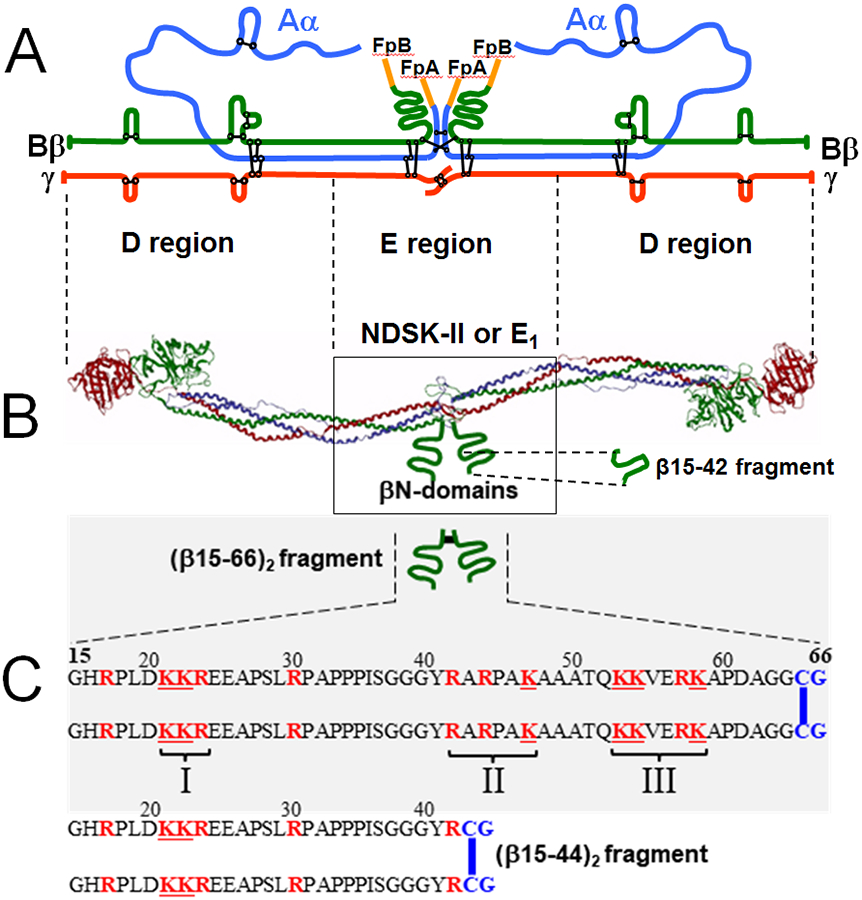
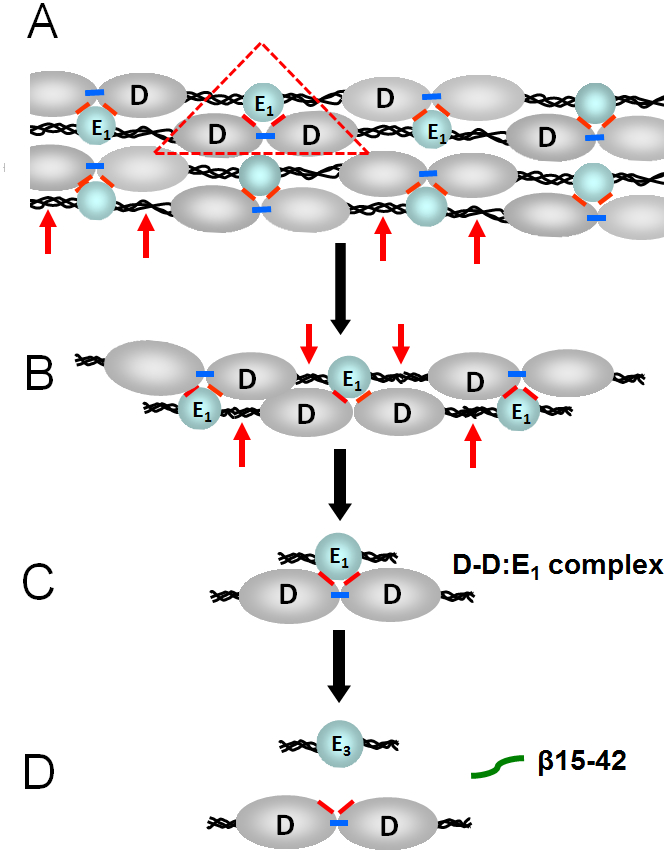
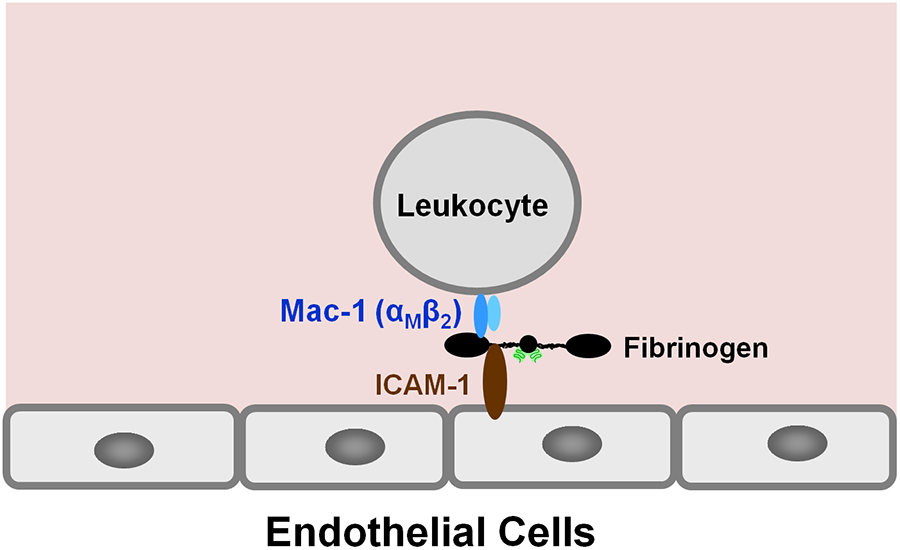
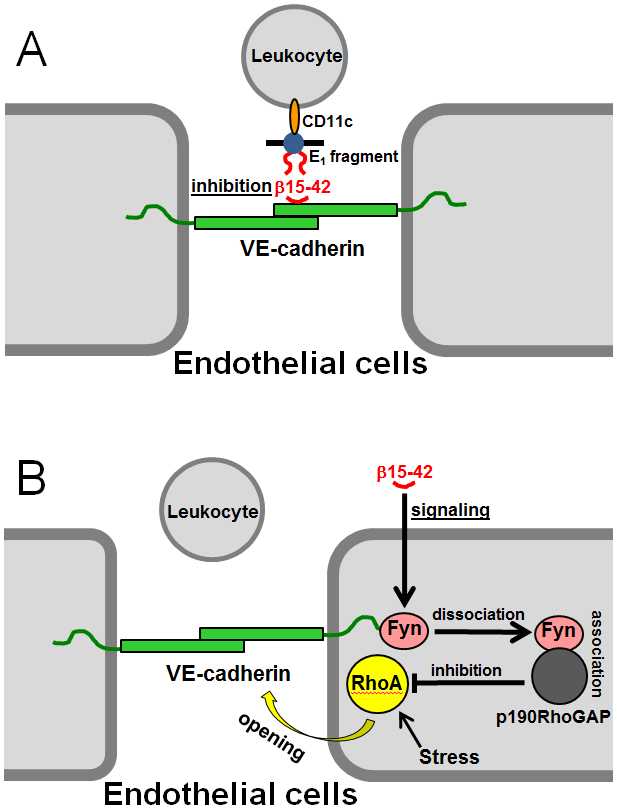
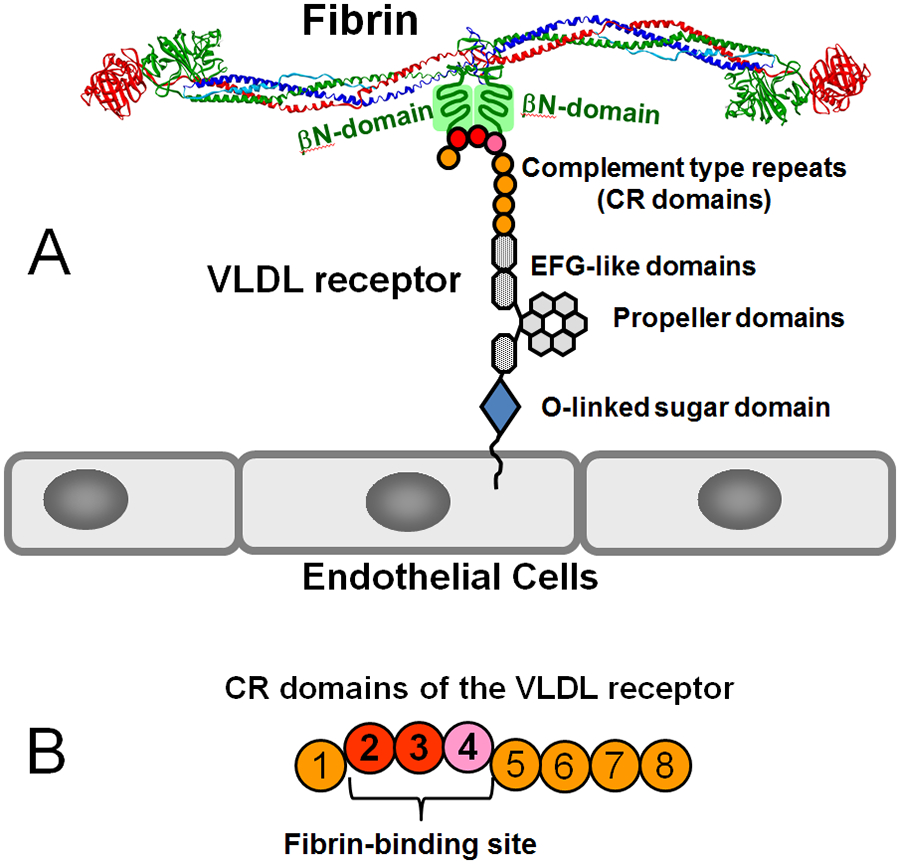
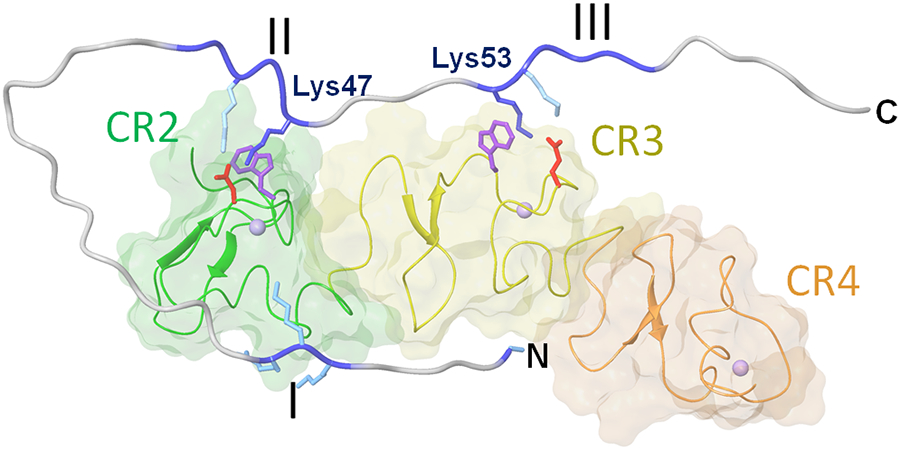
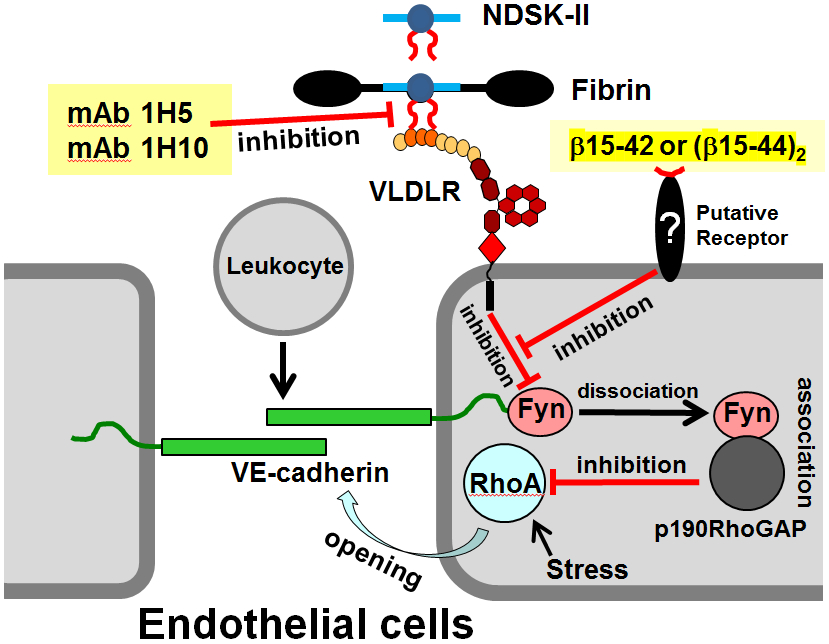
Numerous studies have revealed the involvement of fibrinogen in the inflammatory response. To explain the molecular mechanisms underlying fibrinogen-dependent inflammation, two bridging mechanisms have been proposed in which fibrin(ogen) bridges leukocytes to endothelial cells. The first mechanism suggests that bridging occurs via the interaction of fibrinogen with the leukocyte receptor Mac-1 and the endothelial receptor ICAM-1 (intercellular adhesion molecule-1), which promotes leukocyte transmigration and enhances inflammation. The second mechanism includes bridging of leukocytes to the endothelium by fibrin degradation product E1 fragment through its interaction with leukocyte receptor CD11c and endothelial VE-cadherin to promote leukocyte transmigration. The role of E1 in promoting inflammation is inhibited by the fibrin-derived β15-42 fragment, and this has been suggested to result from its ability to compete for the E1-VE-cadherin interaction and to trigger signaling pathways through the src kinase Fyn. Our recent study revealed that the β15-42 fragment is ineffective in inhibiting the E1- or fibrin-VE-cadherin interaction, leaving the proposed signaling mechanism as the only viable explanation for the inhibitory function of β15-42. We have discovered that fibrin interacts with the very-low-density lipoprotein (VLDL) receptor, and this interaction triggers a signaling pathway that promotes leukocyte transmigration through inhibition of the src kinase Fyn. This pathway is inhibited by another pathway induced by the interaction of β15-42 with a putative endothelial receptor. In this review, we briefly describe the previously proposed molecular mechanisms underlying fibrin-dependent inflammation and their advantages/disadvantages and summarize our recent studies of the novel VLDL receptor-dependent pathway of leukocyte transmigration which plays an important role in fibrin-dependent inflammation.
Thieme. All rights reserved.
Conflict of interest statement
None declared.
Figures
References
-
- Riddle JM, Barnhart ML. The eosinophil as a source for profibrinolysin in acute inflammation. Blood 1965;25(5):776–794 - PubMed
-
- McRitchie DI, Girotti MJ, Glynn MF, Goldberg JM, Rotstein OD. Effect of systemic fibrinogen depletion on intraabdominal abscess formation. J Lab Clin Med 1991;118(1):48–55 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous