

PI3K drives the de novo synthesis of coenzyme A from vitamin B5
- PMID: 35896750
- PMCID: PMC9352595
- DOI: 10.1038/s41586-022-04984-8
PI3K drives the de novo synthesis of coenzyme A from vitamin B5
Abstract
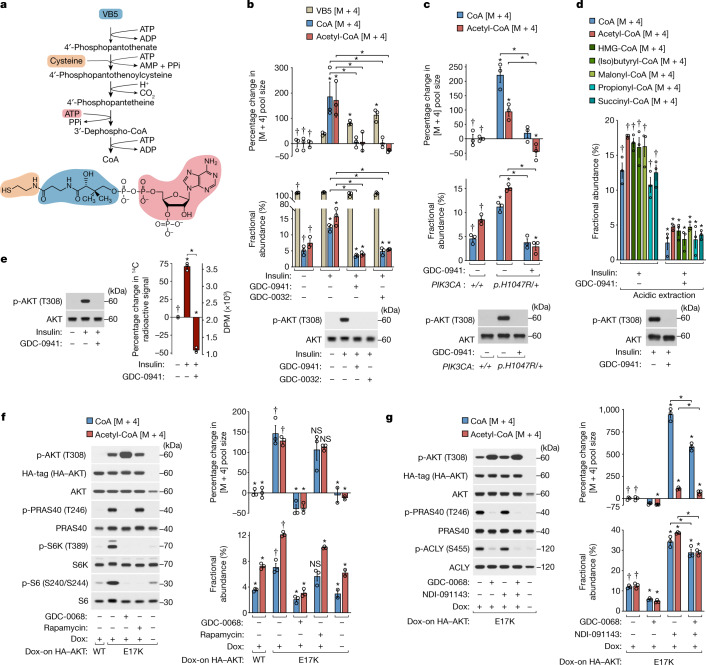
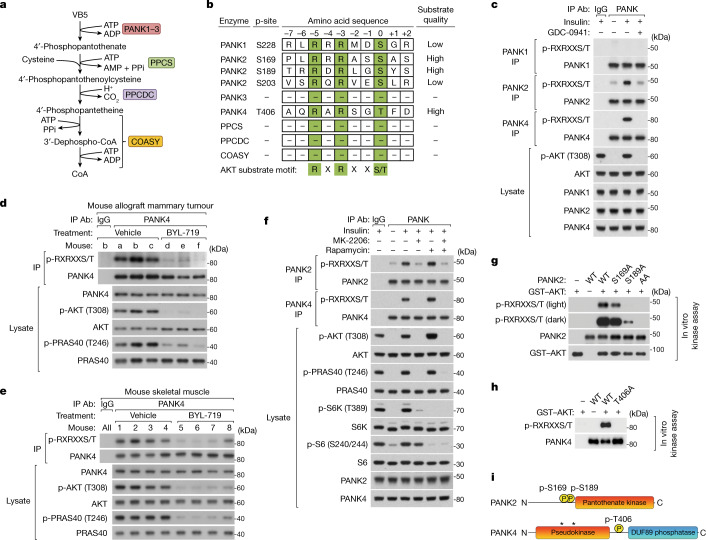
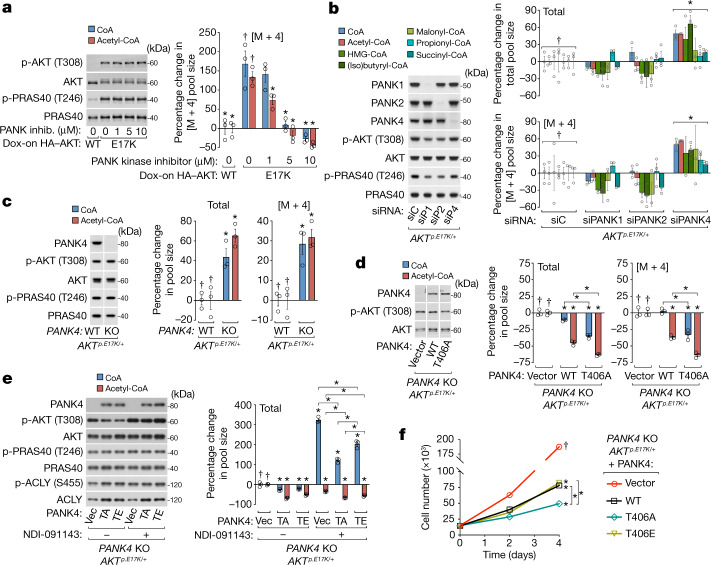
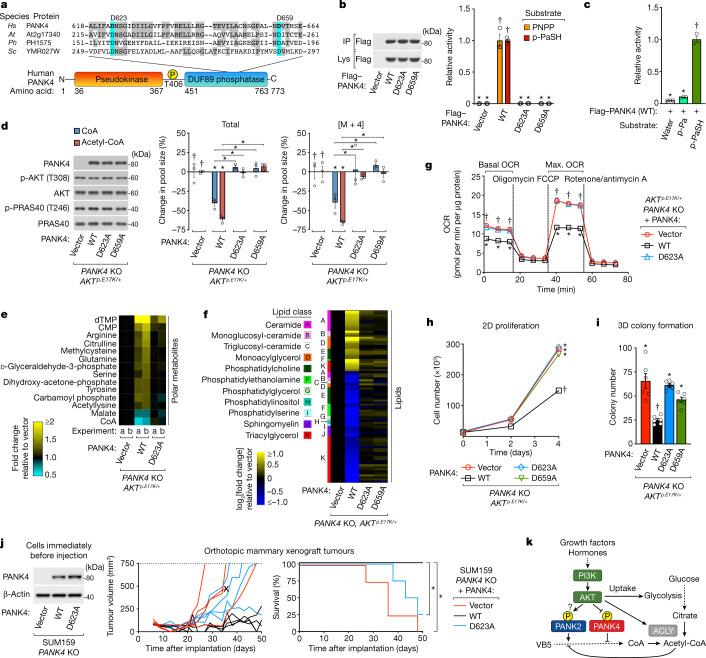
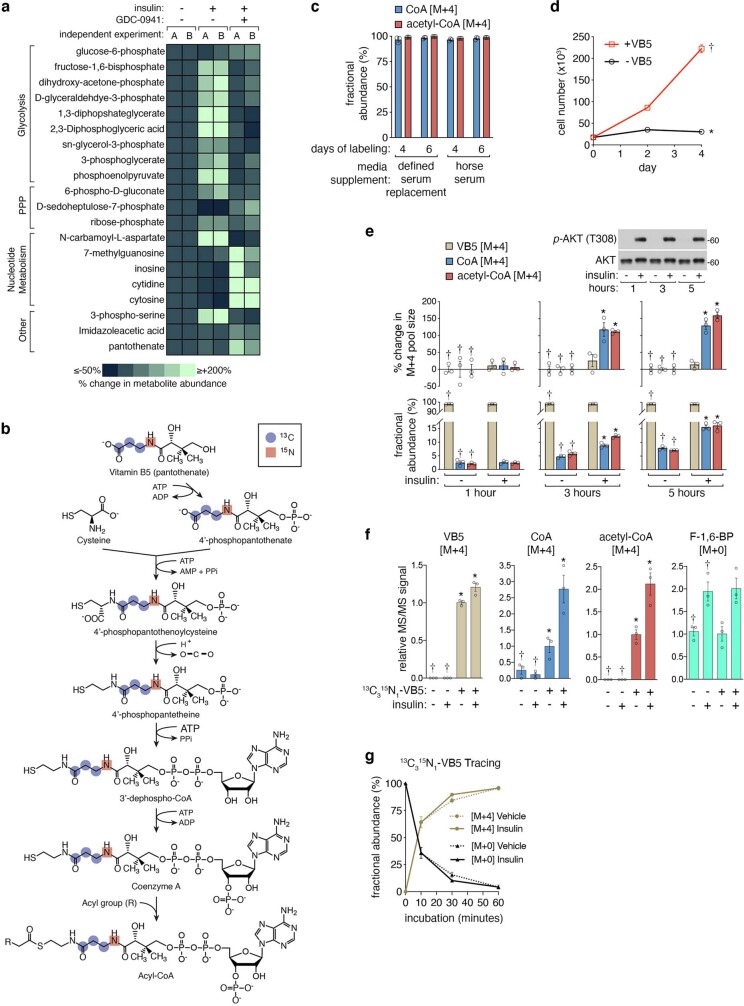
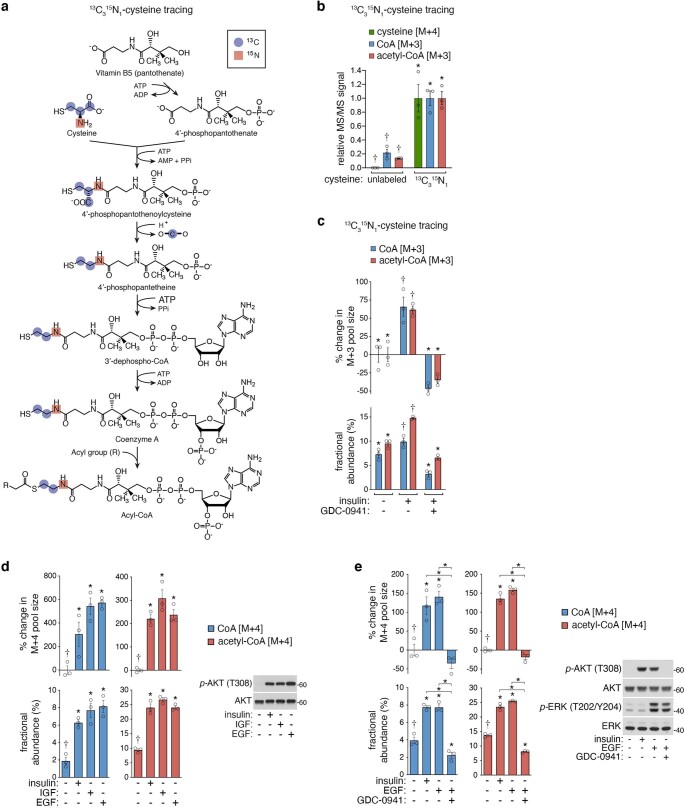
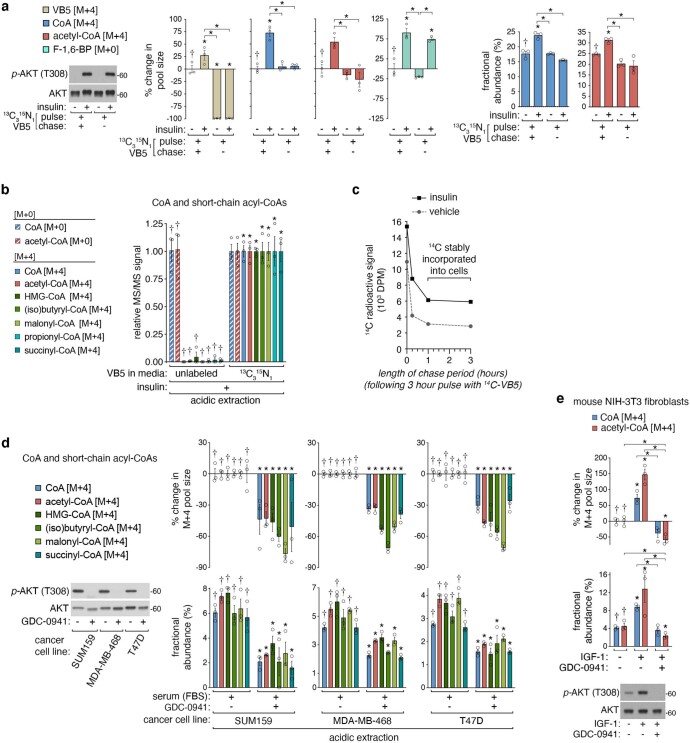
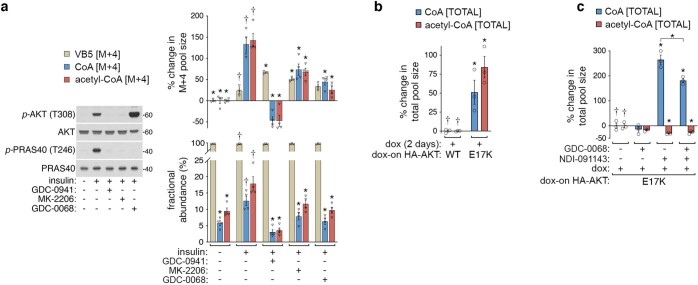
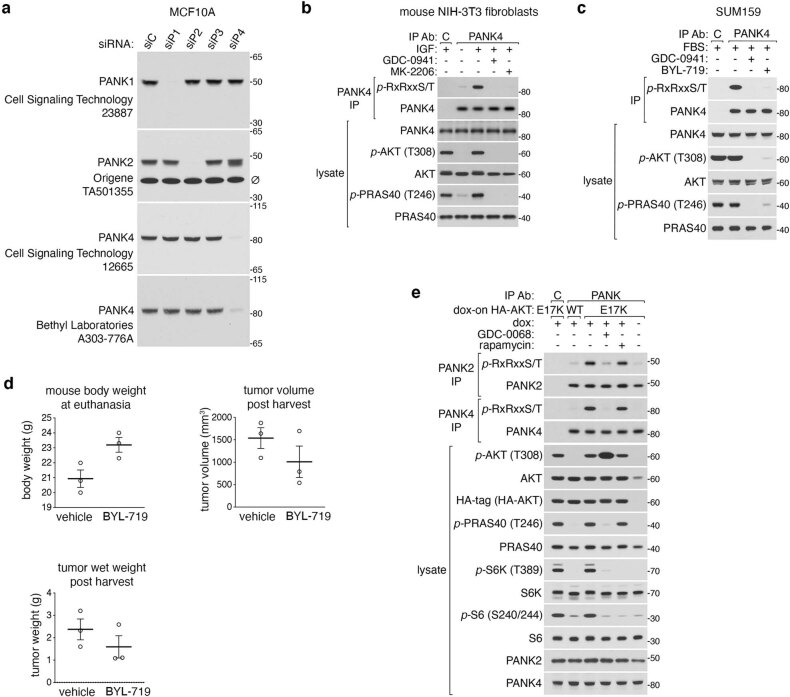
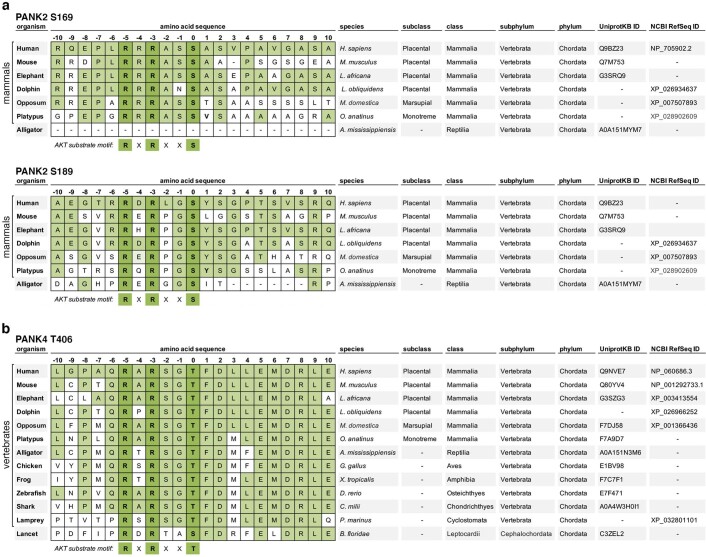
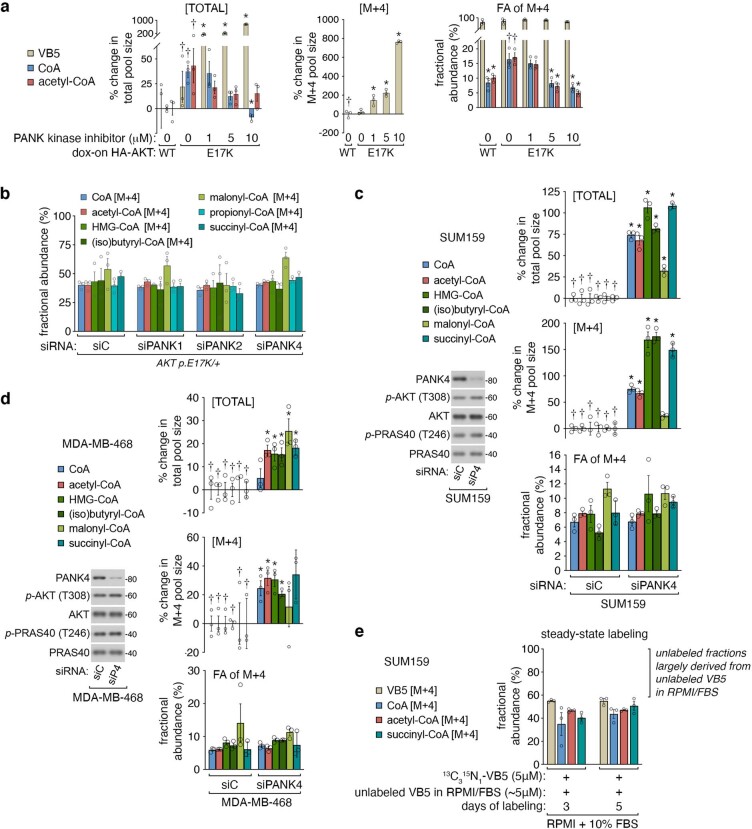
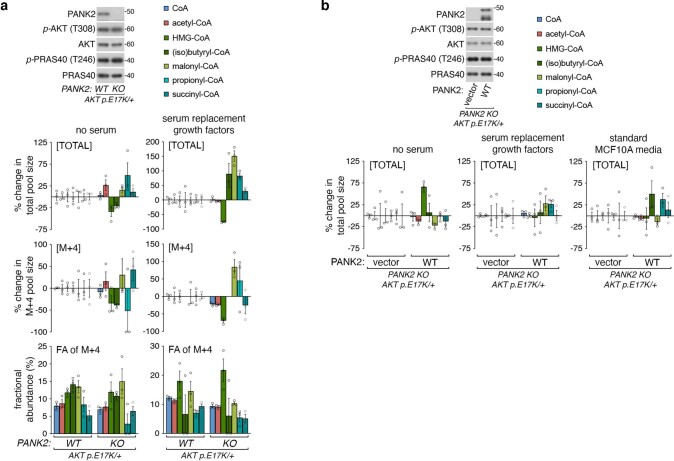
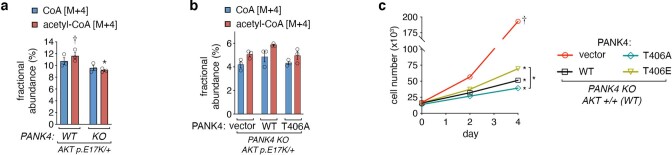
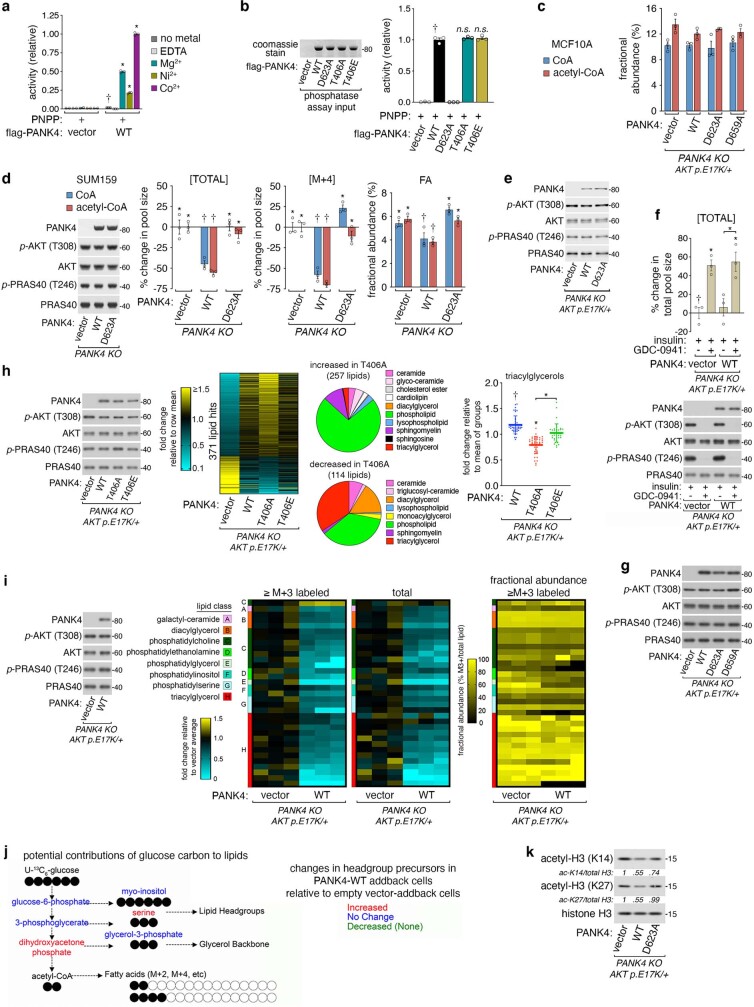
In response to hormones and growth factors, the class I phosphoinositide-3-kinase (PI3K) signalling network functions as a major regulator of metabolism and growth, governing cellular nutrient uptake, energy generation, reducing cofactor production and macromolecule biosynthesis1. Many of the driver mutations in cancer with the highest recurrence, including in receptor tyrosine kinases, Ras, PTEN and PI3K, pathologically activate PI3K signalling2,3. However, our understanding of the core metabolic program controlled by PI3K is almost certainly incomplete. Here, using mass-spectrometry-based metabolomics and isotope tracing, we show that PI3K signalling stimulates the de novo synthesis of one of the most pivotal metabolic cofactors: coenzyme A (CoA). CoA is the major carrier of activated acyl groups in cells4,5 and is synthesized from cysteine, ATP and the essential nutrient vitamin B5 (also known as pantothenate)6,7. We identify pantothenate kinase 2 (PANK2) and PANK4 as substrates of the PI3K effector kinase AKT8. Although PANK2 is known to catalyse the rate-determining first step of CoA synthesis, we find that the minimally characterized but highly conserved PANK49 is a rate-limiting suppressor of CoA synthesis through its metabolite phosphatase activity. Phosphorylation of PANK4 by AKT relieves this suppression. Ultimately, the PI3K-PANK4 axis regulates the abundance of acetyl-CoA and other acyl-CoAs, CoA-dependent processes such as lipid metabolism and proliferation. We propose that these regulatory mechanisms coordinate cellular CoA supplies with the demands of hormone/growth-factor-driven or oncogene-driven metabolism and growth.
© 2022. The Author(s).
Conflict of interest statement
L.C.C. is a founder and scientific advisory board member of Agios Pharmaceuticals, Faeth Therapeutics, Petra Pharma Corporation, Larkspur Therapeutics and Volastra Pharmaceuticals, and scientific advisory board member for Scorpion Therapeutics. B.D.H. is a co-founder of and consultant for Faeth Therapeutics.
Figures
References
-
- Pietrocola F, Galluzzi L, Pedro JMB-S, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. 2015;21:805–821. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
