

SARS-CoV-2 lineage dynamics in England from September to November 2021: high diversity of Delta sub-lineages and increased transmissibility of AY.4.2
- PMID: 35896970
- PMCID: PMC9326417
- DOI: 10.1186/s12879-022-07628-4
SARS-CoV-2 lineage dynamics in England from September to November 2021: high diversity of Delta sub-lineages and increased transmissibility of AY.4.2
Abstract
Background: Since the emergence of SARS-CoV-2, evolutionary pressure has driven large increases in the transmissibility of the virus. However, with increasing levels of immunity through vaccination and natural infection the evolutionary pressure will switch towards immune escape. Genomic surveillance in regions of high immunity is crucial in detecting emerging variants that can more successfully navigate the immune landscape.
Methods: We present phylogenetic relationships and lineage dynamics within England (a country with high levels of immunity), as inferred from a random community sample of individuals who provided a self-administered throat and nose swab for rt-PCR testing as part of the REal-time Assessment of Community Transmission-1 (REACT-1) study. During round 14 (9 September-27 September 2021) and 15 (19 October-5 November 2021) lineages were determined for 1322 positive individuals, with 27.1% of those which reported their symptom status reporting no symptoms in the previous month.
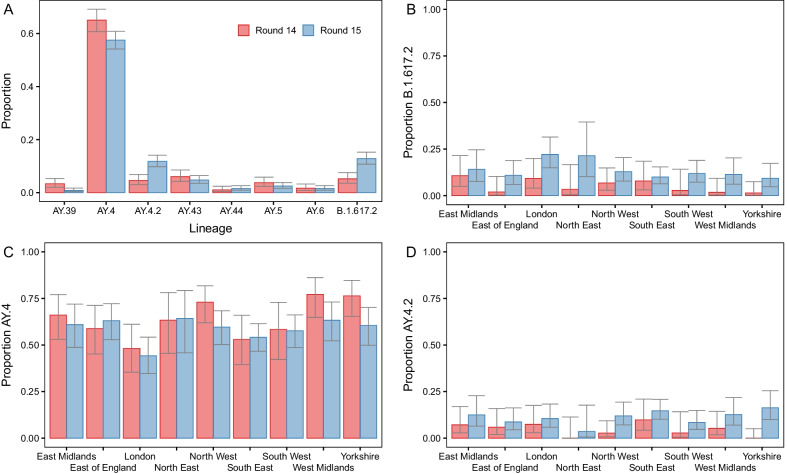
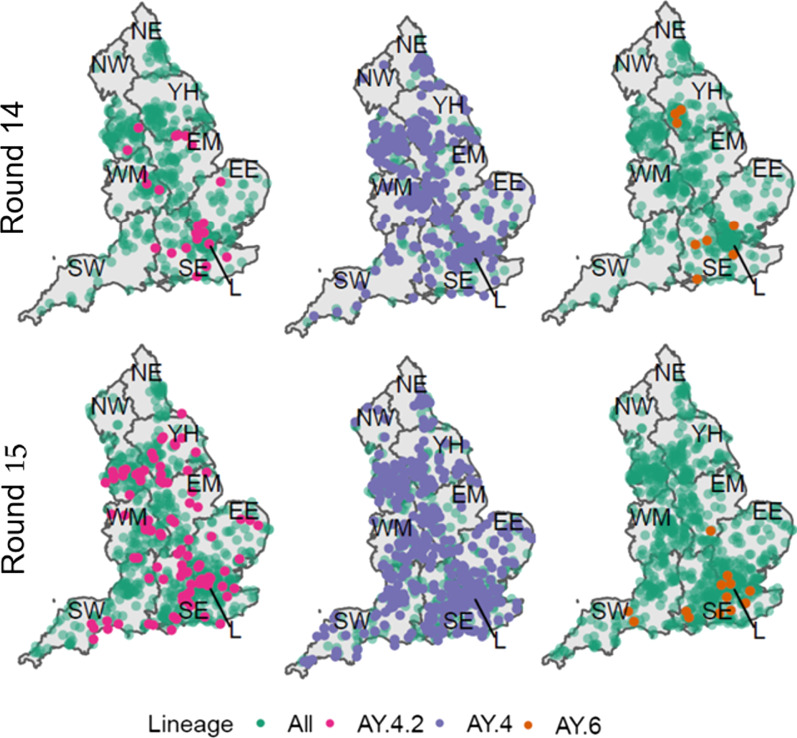
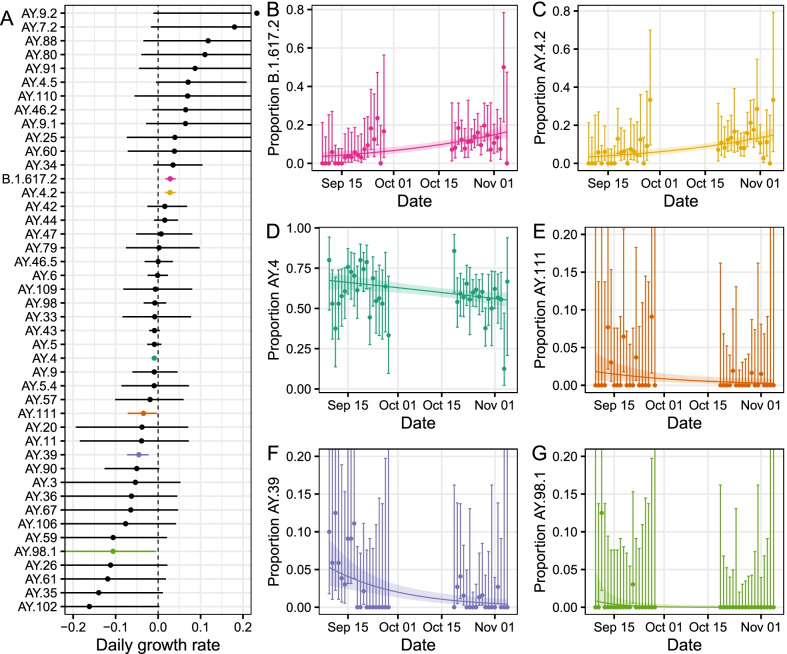
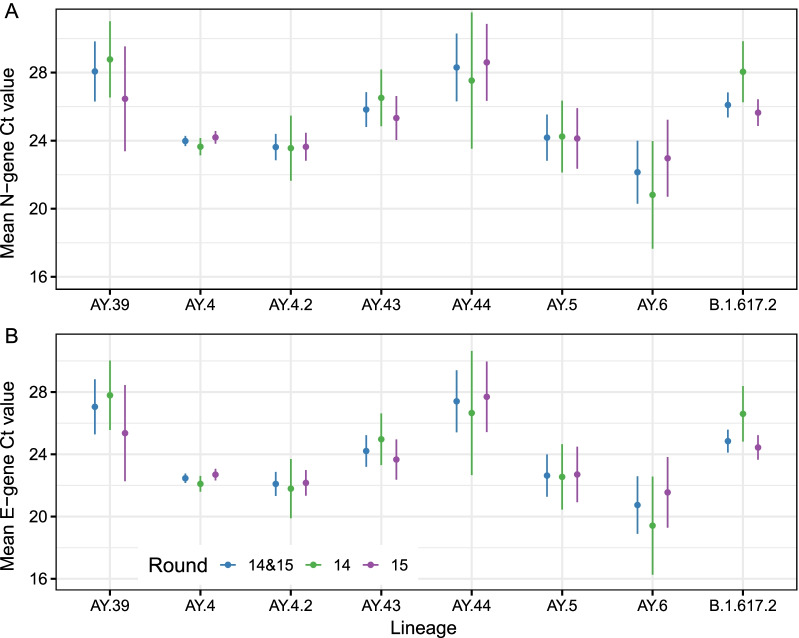
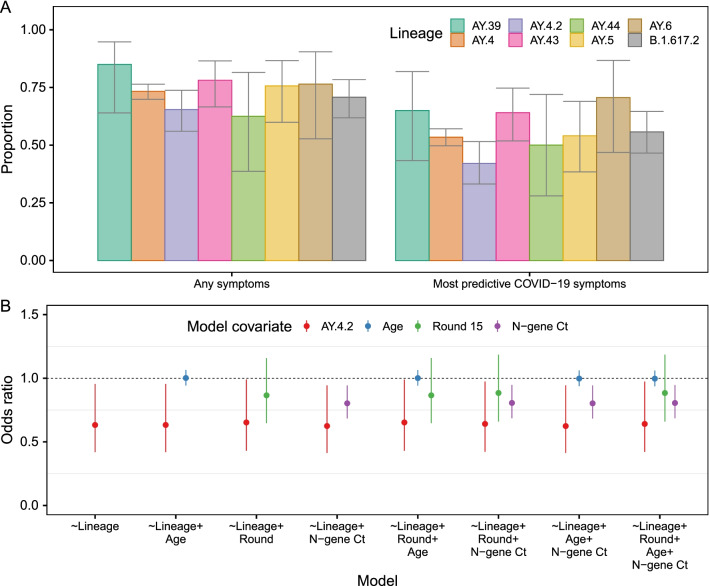
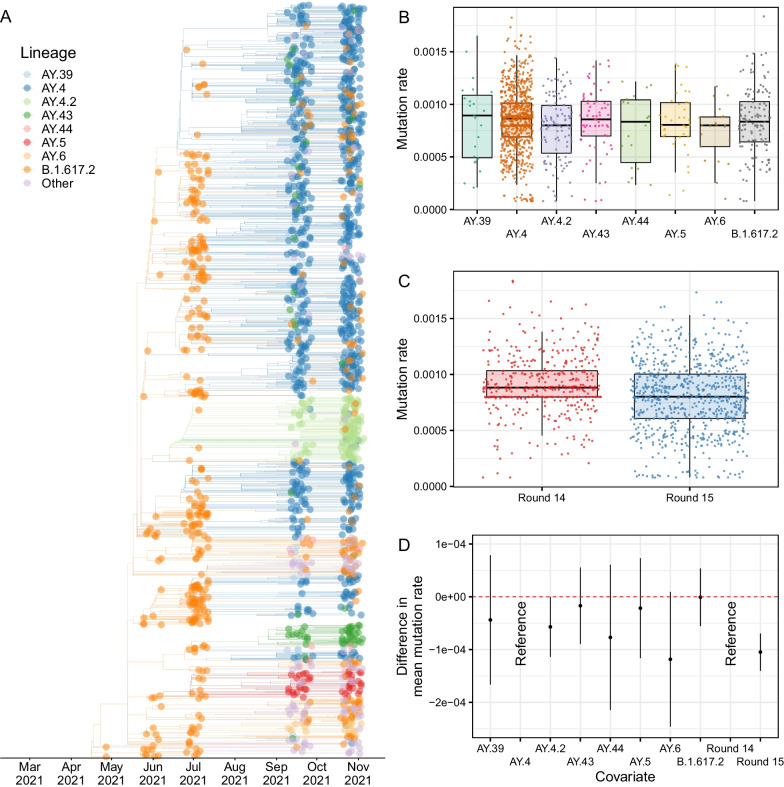
Results: We identified 44 unique lineages, all of which were Delta or Delta sub-lineages, and found a reduction in their mutation rate over the study period. The proportion of the Delta sub-lineage AY.4.2 was increasing, with a reproduction number 15% (95% CI 8-23%) greater than the most prevalent lineage, AY.4. Further, AY.4.2 was less associated with the most predictive COVID-19 symptoms (p = 0.029) and had a reduced mutation rate (p = 0.050). Both AY.4.2 and AY.4 were found to be geographically clustered in September but this was no longer the case by late October/early November, with only the lineage AY.6 exhibiting clustering towards the South of England.
Conclusions: As SARS-CoV-2 moves towards endemicity and new variants emerge, genomic data obtained from random community samples can augment routine surveillance data without the potential biases introduced due to higher sampling rates of symptomatic individuals.
Keywords: COVID-19; Delta variant; Genetic diversity; Mutation; SARS-CoV-2; Transmission advantage.
© 2022. The Author(s).
Conflict of interest statement
We declare no competing interests.
Figures
References
-
- Lineage B.1.617.2 Pangolin report. https://cov-lineages.org/global_report_B.1.617.2.html. Accessed 26 Nov 2021.
-
- World Health Organisation. COVID-19 Weekly Epidemiological Update - Edition 66. 2021. https://www.who.int/publications/m/item/weekly-epidemiological-update-on.... Accessed 23 Nov 2021.
-
- PHE Genomics Cell, PHE Outbreak Surveillance Team, PHE Epidemiology Cell, PHE Contact Tracing Data Team, PHE Health, Protection Data Science Team, PHE Joint Modelling Team, NHS Test and Trace Joint Biosecurity Centre, Public Health Scotland and EAVE group, Contributions from the Variant Technical Group Members. SARS-CoV-2 variants of concern and variants under investigation in England - Technical briefing 15, 11 June 2021. https://assets.publishing.service.gov.uk/government/uploads/system/uploa...
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
