

miR-489 Confines Uncontrolled Estrogen Signaling through a Negative Feedback Mechanism and Regulates Tamoxifen Resistance in Breast Cancer
- PMID: 35897675
- PMCID: PMC9331933
- DOI: 10.3390/ijms23158086
miR-489 Confines Uncontrolled Estrogen Signaling through a Negative Feedback Mechanism and Regulates Tamoxifen Resistance in Breast Cancer
Abstract
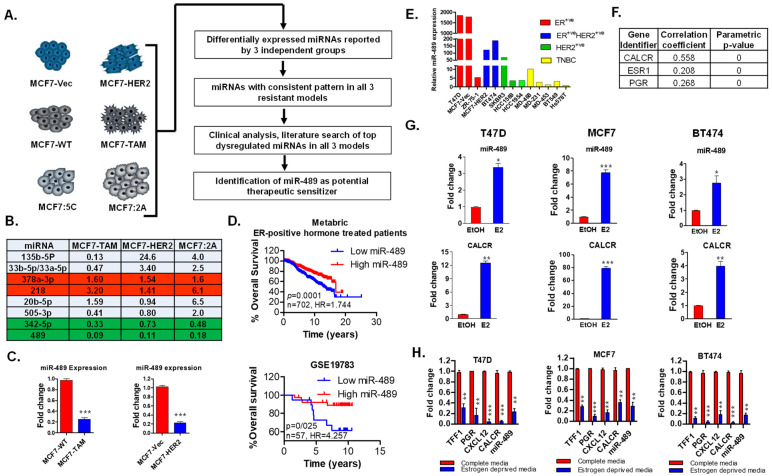
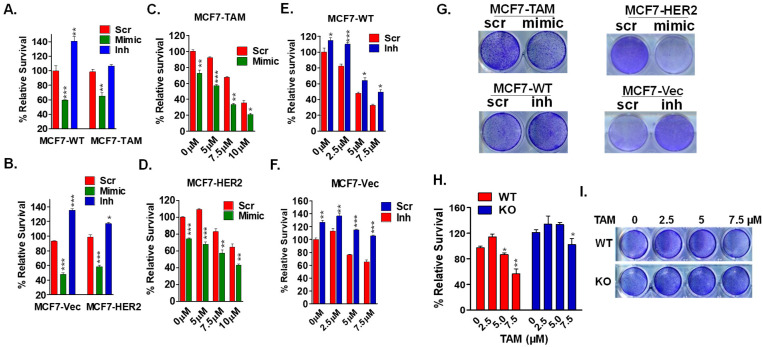
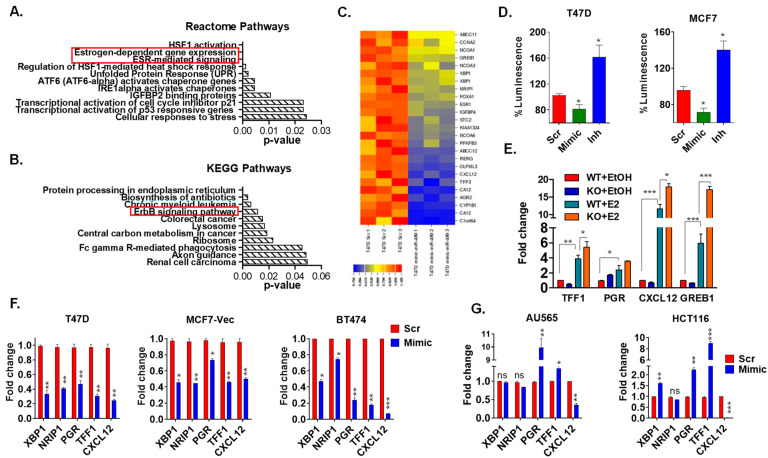
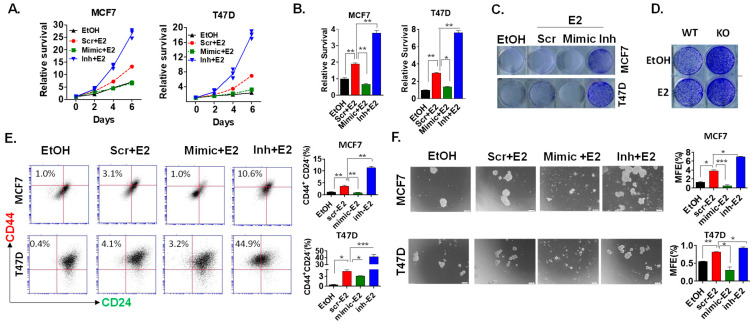
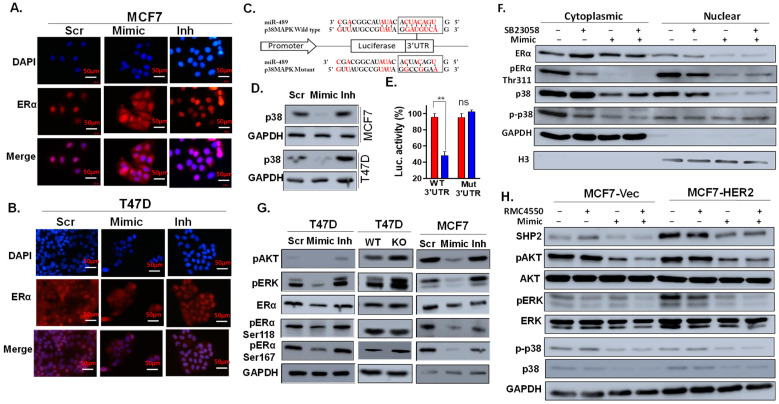
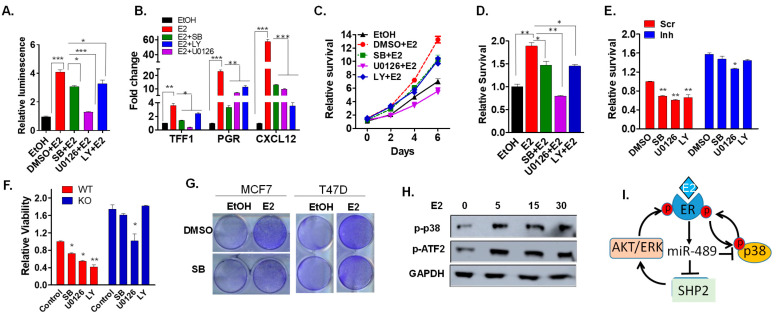
Approximately 75% of diagnosed breast cancer tumors are estrogen-receptor-positive tumors and are associated with a better prognosis due to response to hormonal therapies. However, around 40% of patients relapse after hormonal therapies. Genomic analysis of gene expression profiles in primary breast cancers and tamoxifen-resistant cell lines suggested the potential role of miR-489 in the regulation of estrogen signaling and development of tamoxifen resistance. Our in vitro analysis showed that loss of miR-489 expression promoted tamoxifen resistance, while overexpression of miR-489 in tamoxifen-resistant cells restored tamoxifen sensitivity. Mechanistically, we found that miR-489 is an estrogen-regulated miRNA that negatively regulates estrogen receptor signaling by using at least the following two mechanisms: (i) modulation of the ER phosphorylation status by inhibiting MAPK and AKT kinase activities; (ii) regulation of nuclear-to-cytosol translocation of estrogen receptor α (ERα) by decreasing p38 expression and consequently ER phosphorylation. In addition, miR-489 can break the positive feed-forward loop between the estrogen-Erα axis and p38 MAPK in breast cancer cells, which is necessary for its function as a transcription factor. Overall, our study unveiled the underlying molecular mechanism by which miR-489 regulates an estrogen signaling pathway through a negative feedback loop and uncovered its role in both the development of and overcoming of tamoxifen resistance in breast cancers.
Keywords: CRISPR/Cas9; breast cancer; estrogen receptor; miR-489; tamoxifen resistance.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
