

The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention
- PMID: 35897717
- PMCID: PMC9331760
- DOI: 10.3390/ijms23158141
The Role of HDACs in the Response of Cancer Cells to Cellular Stress and the Potential for Therapeutic Intervention
Abstract
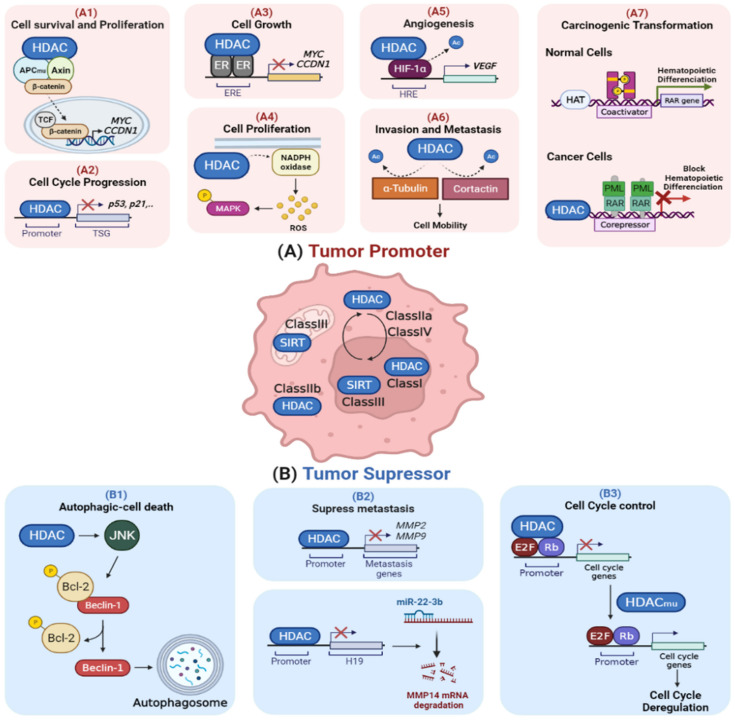
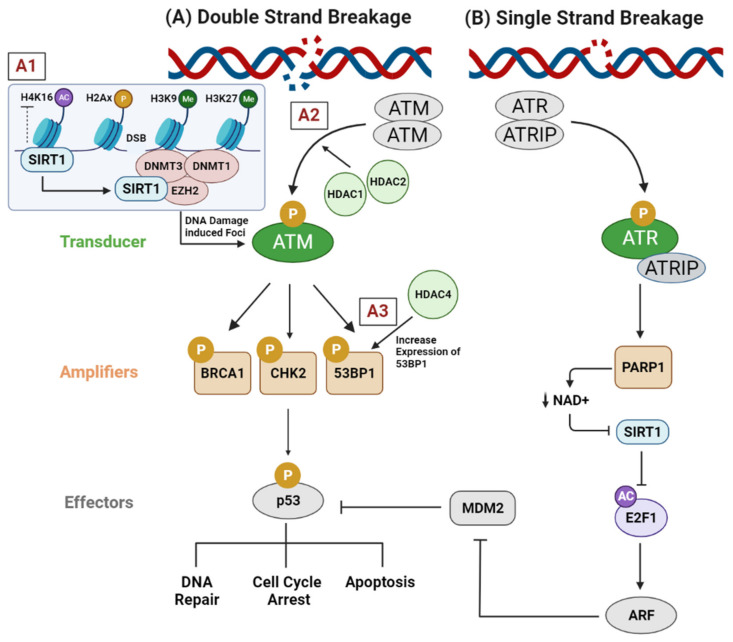
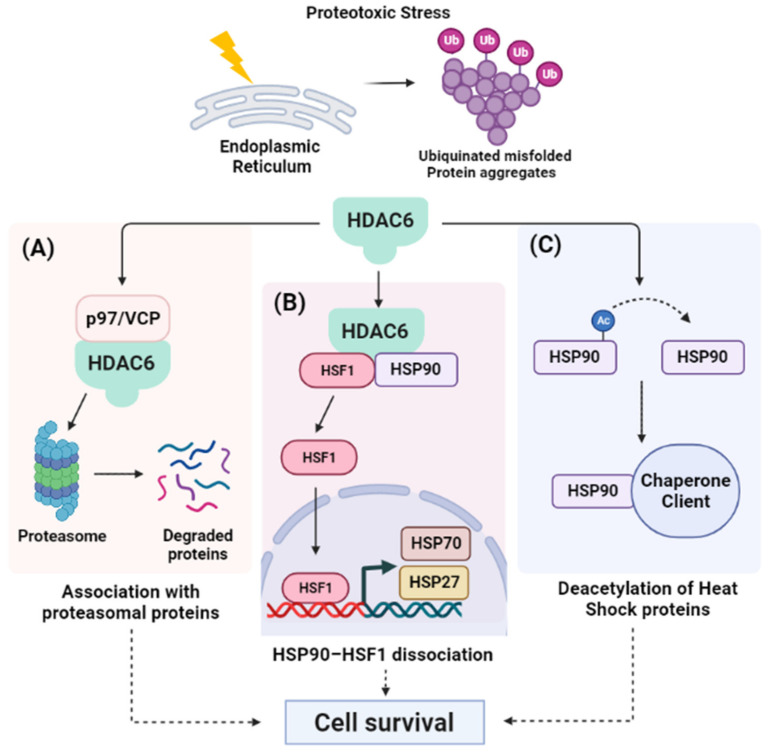
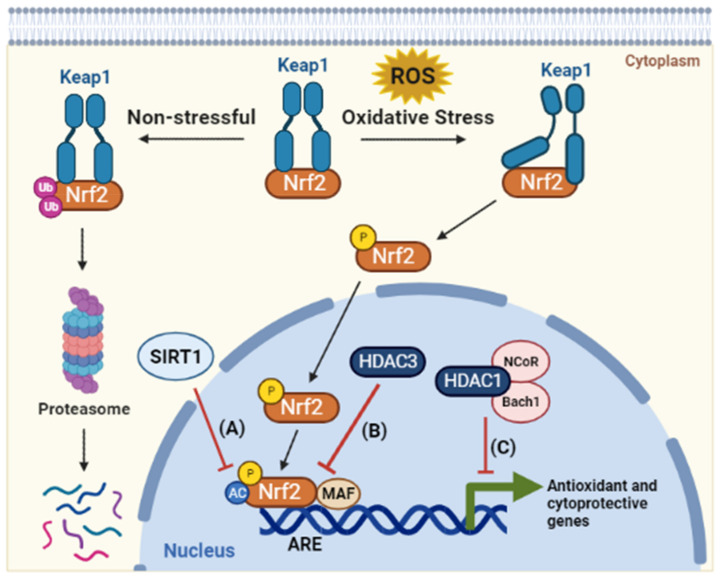
Throughout the process of carcinogenesis, cancer cells develop intricate networks to adapt to a variety of stressful conditions including DNA damage, nutrient deprivation, and hypoxia. These molecular networks encounter genomic instability and mutations coupled with changes in the gene expression programs due to genetic and epigenetic alterations. Histone deacetylases (HDACs) are important modulators of the epigenetic constitution of cancer cells. It has become increasingly known that HDACs have the capacity to regulate various cellular systems through the deacetylation of histone and bounteous nonhistone proteins that are rooted in complex pathways in cancer cells to evade death pathways and immune surveillance. Elucidation of the signaling pathways involved in the adaptive responses to cellular stress and the role of HDACs may lead to the development of novel therapeutic agents. In this article, we overview the dominant stress types including metabolic, oxidative, genotoxic, and proteotoxic stress imposed on cancer cells in the context of HDACs, which guide stress adaptation responses. Next, we expose a closer view on the therapeutic interventions and clinical trials that involve HDACs inhibitors, in addition to highlighting the impact of using HDAC inhibitors in combination with stress-inducing agents for the management of cancer and to overcome the resistance to current cancer therapy.
Keywords: HDAC inhibitors; HDACs; cellular stress; immune modulation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
