

Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy
- PMID: 35897913
- PMCID: PMC9330692
- DOI: 10.3390/molecules27154736
Targeting Replication Stress Response Pathways to Enhance Genotoxic Chemo- and Radiotherapy
Abstract
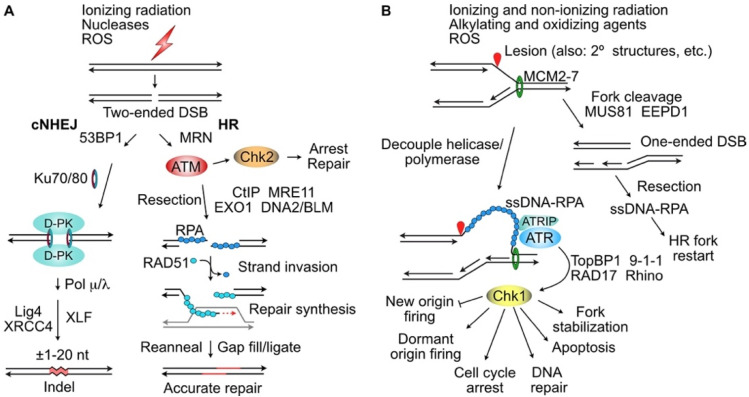
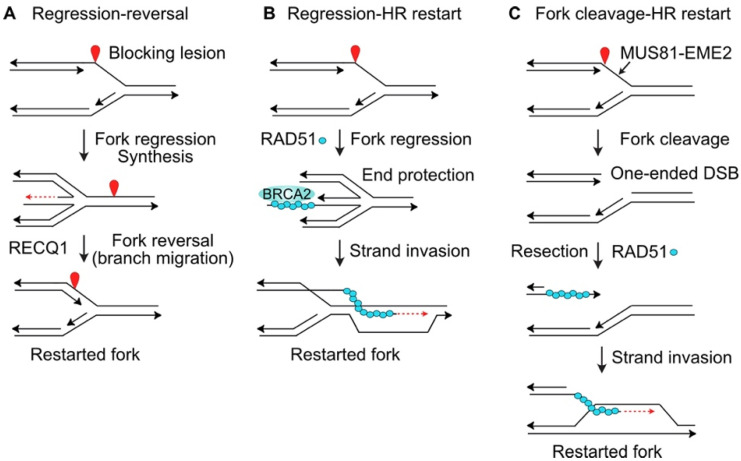
Proliferating cells regularly experience replication stress caused by spontaneous DNA damage that results from endogenous reactive oxygen species (ROS), DNA sequences that can assume secondary and tertiary structures, and collisions between opposing transcription and replication machineries. Cancer cells face additional replication stress, including oncogenic stress that results from the dysregulation of fork progression and origin firing, and from DNA damage induced by radiotherapy and most cancer chemotherapeutic agents. Cells respond to such stress by activating a complex network of sensor, signaling and effector pathways that protect genome integrity. These responses include slowing or stopping active replication forks, protecting stalled replication forks from collapse, preventing late origin replication firing, stimulating DNA repair pathways that promote the repair and restart of stalled or collapsed replication forks, and activating dormant origins to rescue adjacent stressed forks. Currently, most cancer patients are treated with genotoxic chemotherapeutics and/or ionizing radiation, and cancer cells can gain resistance to the resulting replication stress by activating pro-survival replication stress pathways. Thus, there has been substantial effort to develop small molecule inhibitors of key replication stress proteins to enhance tumor cell killing by these agents. Replication stress targets include ATR, the master kinase that regulates both normal replication and replication stress responses; the downstream signaling kinase Chk1; nucleases that process stressed replication forks (MUS81, EEPD1, Metnase); the homologous recombination catalyst RAD51; and other factors including ATM, DNA-PKcs, and PARP1. This review provides an overview of replication stress response pathways and discusses recent pre-clinical studies and clinical trials aimed at improving cancer therapy by targeting replication stress response factors.
Keywords: DNA damage response; DNA replication stress; genotoxic cancer therapy; oncogenic stress; targeted therapy.
Conflict of interest statement
The author declares no conflict of interest.
Figures
Similar articles
-
Cellular Responses to Widespread DNA Replication Stress.Int J Mol Sci. 2023 Nov 29;24(23):16903. doi: 10.3390/ijms242316903. Int J Mol Sci. 2023. PMID: 38069223 Free PMC article. Review.
-
EEPD1 Rescues Stressed Replication Forks and Maintains Genome Stability by Promoting End Resection and Homologous Recombination Repair.PLoS Genet. 2015 Dec 18;11(12):e1005675. doi: 10.1371/journal.pgen.1005675. eCollection 2015 Dec. PLoS Genet. 2015. PMID: 26684013 Free PMC article.
-
Distinct roles of structure-specific endonucleases EEPD1 and Metnase in replication stress responses.NAR Cancer. 2020 Jun;2(2):zcaa008. doi: 10.1093/narcan/zcaa008. Epub 2020 Jun 8. NAR Cancer. 2020. PMID: 32743552 Free PMC article.
-
Metnase and EEPD1: DNA Repair Functions and Potential Targets in Cancer Therapy.Front Oncol. 2022 Jan 28;12:808757. doi: 10.3389/fonc.2022.808757. eCollection 2022. Front Oncol. 2022. PMID: 35155245 Free PMC article. Review.
-
Nucleases and Co-Factors in DNA Replication Stress Responses.DNA (Basel). 2022 Mar;2(1):68-85. doi: 10.3390/dna2010006. Epub 2022 Mar 1. DNA (Basel). 2022. PMID: 36203968 Free PMC article.
Cited by
-
The Multiple Faces of the MRN Complex: Roles in Medulloblastoma and Beyond.Cancers (Basel). 2023 Jul 13;15(14):3599. doi: 10.3390/cancers15143599. Cancers (Basel). 2023. PMID: 37509263 Free PMC article. Review.
-
New Facets of DNA Double Strand Break Repair: Radiation Dose as Key Determinant of HR versus c-NHEJ Engagement.Int J Mol Sci. 2023 Oct 6;24(19):14956. doi: 10.3390/ijms241914956. Int J Mol Sci. 2023. PMID: 37834403 Free PMC article. Review.
-
Prognostic Assessment of Oxidative Stress-Related Genes in Colorectal Cancer and New Insights into Tumor Immunity.Oxid Med Cell Longev. 2022 Oct 15;2022:2518340. doi: 10.1155/2022/2518340. eCollection 2022. Oxid Med Cell Longev. 2022. PMID: 36299603 Free PMC article.
-
Tumor acidosis-induced DNA damage response and tetraploidy enhance sensitivity to ATM and ATR inhibitors.EMBO Rep. 2024 Mar;25(3):1469-1489. doi: 10.1038/s44319-024-00089-7. Epub 2024 Feb 16. EMBO Rep. 2024. PMID: 38366255 Free PMC article.
-
Cellular Responses to Widespread DNA Replication Stress.Int J Mol Sci. 2023 Nov 29;24(23):16903. doi: 10.3390/ijms242316903. Int J Mol Sci. 2023. PMID: 38069223 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
