

Uniform binding and negative catalysis at the origin of enzymes
- PMID: 35900021
- PMCID: PMC9281367
- DOI: 10.1002/pro.4381
Uniform binding and negative catalysis at the origin of enzymes
Abstract
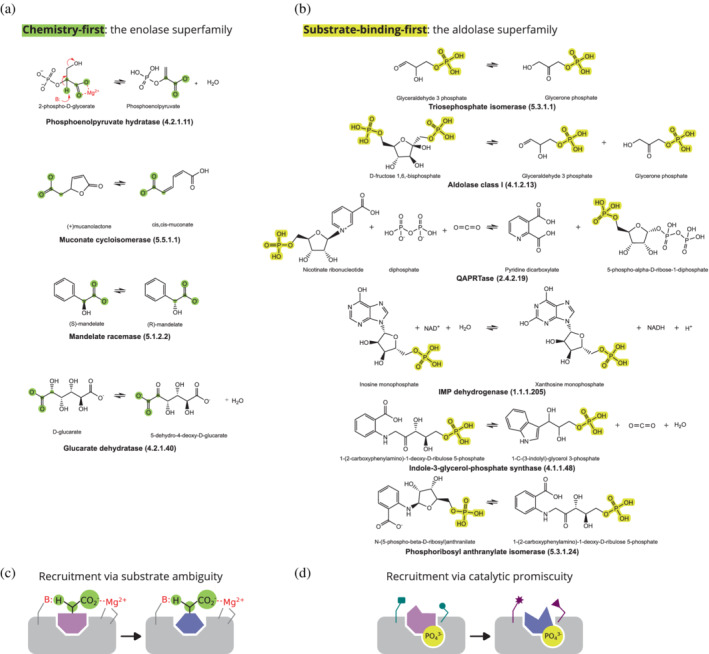
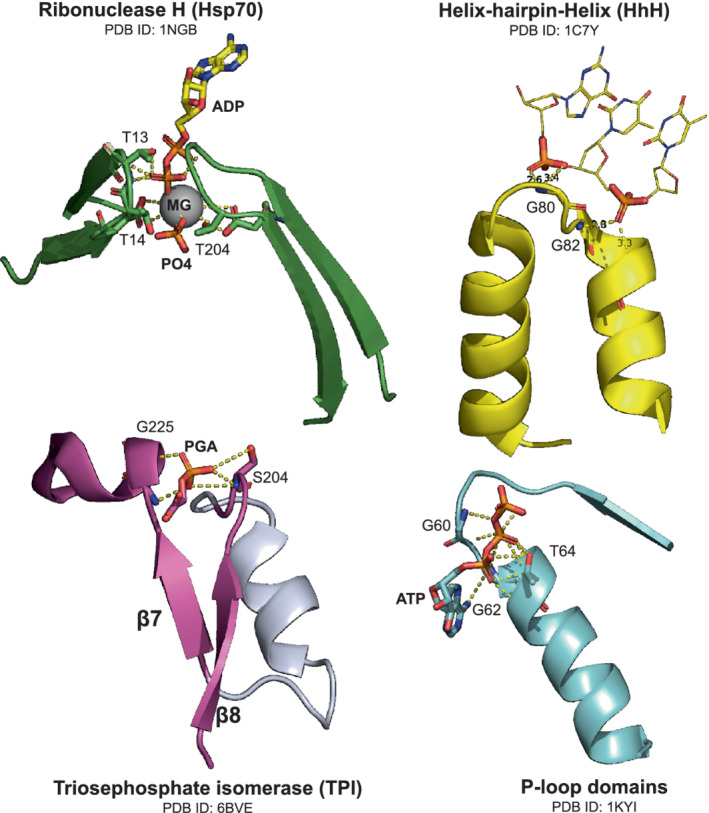
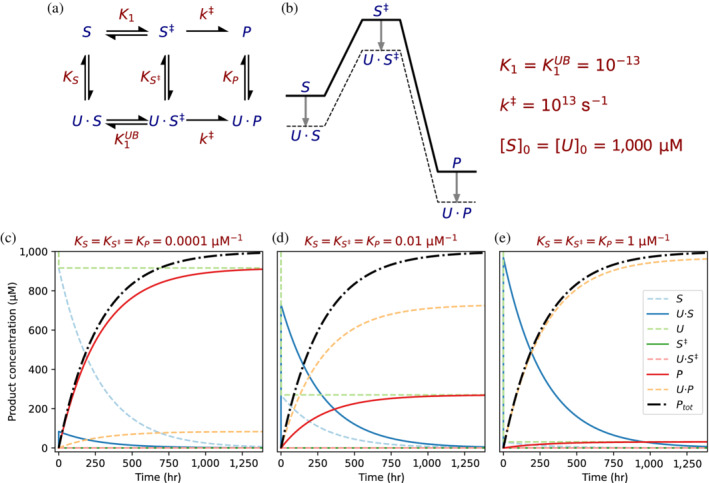
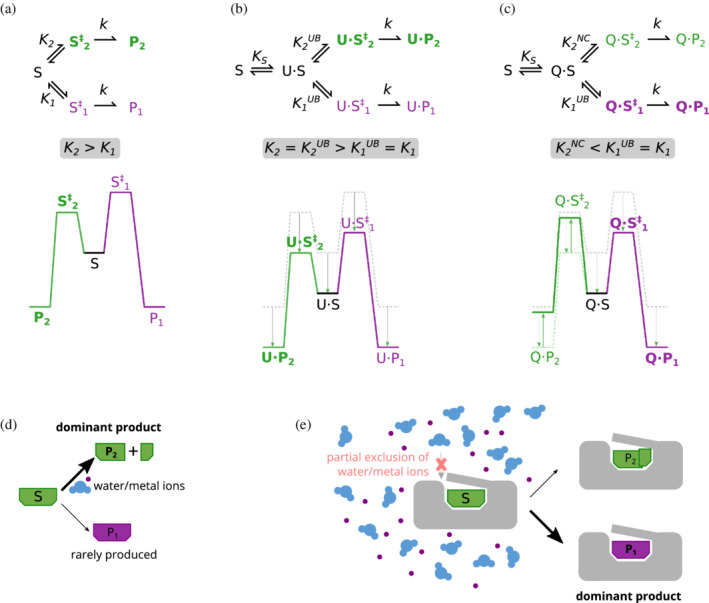
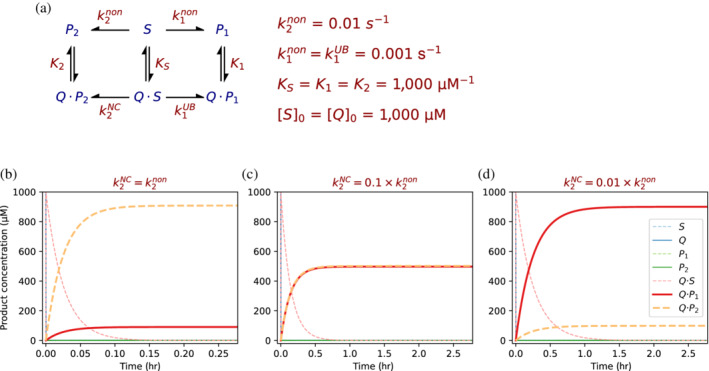
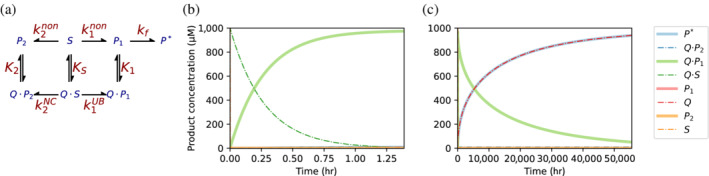
Enzymes are well known for their catalytic abilities, some even reaching "catalytic perfection" in the sense that the reaction they catalyze has reached the physical bound of the diffusion rate. However, our growing understanding of enzyme superfamilies has revealed that only some share a catalytic chemistry while others share a substrate-handle binding motif, for example, for a particular phosphate group. This suggests that some families emerged through a "substrate-handle-binding-first" mechanism ("binding-first" for brevity) instead of "chemistry-first" and we are, therefore, left to wonder what the role of non-catalytic binders might have been during enzyme evolution. In the last of their eight seminal, back-to-back articles from 1976, John Albery and Jeremy Knowles addressed the question of enzyme evolution by arguing that the simplest mode of enzyme evolution is what they defined as "uniform binding" (parallel stabilization of all enzyme-bound states to the same degree). Indeed, we show that a uniform-binding proto-catalyst can accelerate a reaction, but only when catalysis is already present, that is, when the transition state is already stabilized to some degree. Thus, we sought an alternative explanation for the cases where substrate-handle-binding preceded any involvement of a catalyst. We find that evolutionary starting points that exhibit negative catalysis can redirect the reaction's course to a preferred product without need for rate acceleration or product release; that is, if they do not stabilize, or even destabilize, the transition state corresponding to an undesired product. Such a mechanism might explain the emergence of "binding-first" enzyme families like the aldolase superfamily.
Keywords: enzyme evolution; negative catalysis; primordial catalyst; substrate-handle; triose phosphate isomerase; uniform binding.
© 2022 The Authors. Protein Science published by Wiley Periodicals LLC on behalf of The Protein Society.
Figures

Similar articles
-
Evolution of Enzyme Function and the Development of Catalytic Efficiency: Triosephosphate Isomerase, Jeremy R. Knowles, and W. John Albery.Biochemistry. 2021 Nov 23;60(46):3529-3538. doi: 10.1021/acs.biochem.1c00211. Epub 2021 May 20. Biochemistry. 2021. PMID: 34015914 Free PMC article.
-
Why do many Michaelian enzymes exhibit an equilibrium constant close to unity for the interconversion of enzyme-bound substrate and product?Eur J Biochem. 1991 Feb 14;195(3):663-70. doi: 10.1111/j.1432-1033.1991.tb15751.x. Eur J Biochem. 1991. PMID: 1999189
-
Enabling Role of Ligand-Driven Conformational Changes in Enzyme Evolution.Biochemistry. 2022 Aug 2;61(15):1533-1542. doi: 10.1021/acs.biochem.2c00178. Epub 2022 Jul 13. Biochemistry. 2022. PMID: 35829700 Free PMC article. Review.
-
Design of biomimetic catalysts by molecular imprinting in synthetic polymers: the role of transition state stabilization.Acc Chem Res. 2012 Feb 21;45(2):239-47. doi: 10.1021/ar200146m. Epub 2011 Oct 3. Acc Chem Res. 2012. PMID: 21967389
-
Role of dynamics in enzyme catalysis: substantial versus semantic controversies.Acc Chem Res. 2015 Feb 17;48(2):466-73. doi: 10.1021/ar500322s. Epub 2014 Dec 24. Acc Chem Res. 2015. PMID: 25539442 Free PMC article. Review.
Cited by
-
Tracing the birth and intrinsic disorder of loops and domains in protein evolution.Biophys Rev. 2024 Nov 20;16(6):723-735. doi: 10.1007/s12551-024-01251-0. eCollection 2024 Dec. Biophys Rev. 2024. PMID: 39830125 Free PMC article. Review.
-
On the Origins of Enzymes: Phosphate-Binding Polypeptides Mediate Phosphoryl Transfer to Synthesize Adenosine Triphosphate.J Am Chem Soc. 2023 Mar 23;145(15):8344-54. doi: 10.1021/jacs.2c08636. Online ahead of print. J Am Chem Soc. 2023. PMID: 36951643 Free PMC article.
-
Reflections on the Origin of Coded Protein Biosynthesis.Biomolecules. 2024 Apr 25;14(5):518. doi: 10.3390/biom14050518. Biomolecules. 2024. PMID: 38785925 Free PMC article. Review.
-
Prebiotic Synthesis of ATP: A Terrestrial Volcanism-Dependent Pathway.Life (Basel). 2023 Mar 8;13(3):731. doi: 10.3390/life13030731. Life (Basel). 2023. PMID: 36983886 Free PMC article.
-
Back in time to the Gly-rich prototype of the phosphate binding elementary function.Curr Res Struct Biol. 2024 Apr 9;7:100142. doi: 10.1016/j.crstbi.2024.100142. eCollection 2024. Curr Res Struct Biol. 2024. PMID: 38655428 Free PMC article. Review.
References
-
- Albery WJ, Knowles JR. Evolution of enzyme function and the development of catalytic efficiency. Biochemistry. 1976;15:5631–5640. - PubMed
-
- Fersht A. Structure and mechanism in protein science: A guide to enzyme catalysis and protein folding. New York: W. H. Freeman, 1999.
-
- Ycas M. On earlier states of the biochemical system. J Theor Biol. 1974;44:145–160. - PubMed
-
- Jensen RA. Enzyme recruitment in evolution of new function. Annu Rev Microbiol. 1976;30:409–425. - PubMed
-
- Jacob F. Evolution and tinkering. Science. 1977;196:1161–1166. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
