

Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment
- PMID: 35902743
- PMCID: PMC10290535
- DOI: 10.1038/s41588-022-01134-8
Single-nucleus and spatial transcriptome profiling of pancreatic cancer identifies multicellular dynamics associated with neoadjuvant treatment
Abstract
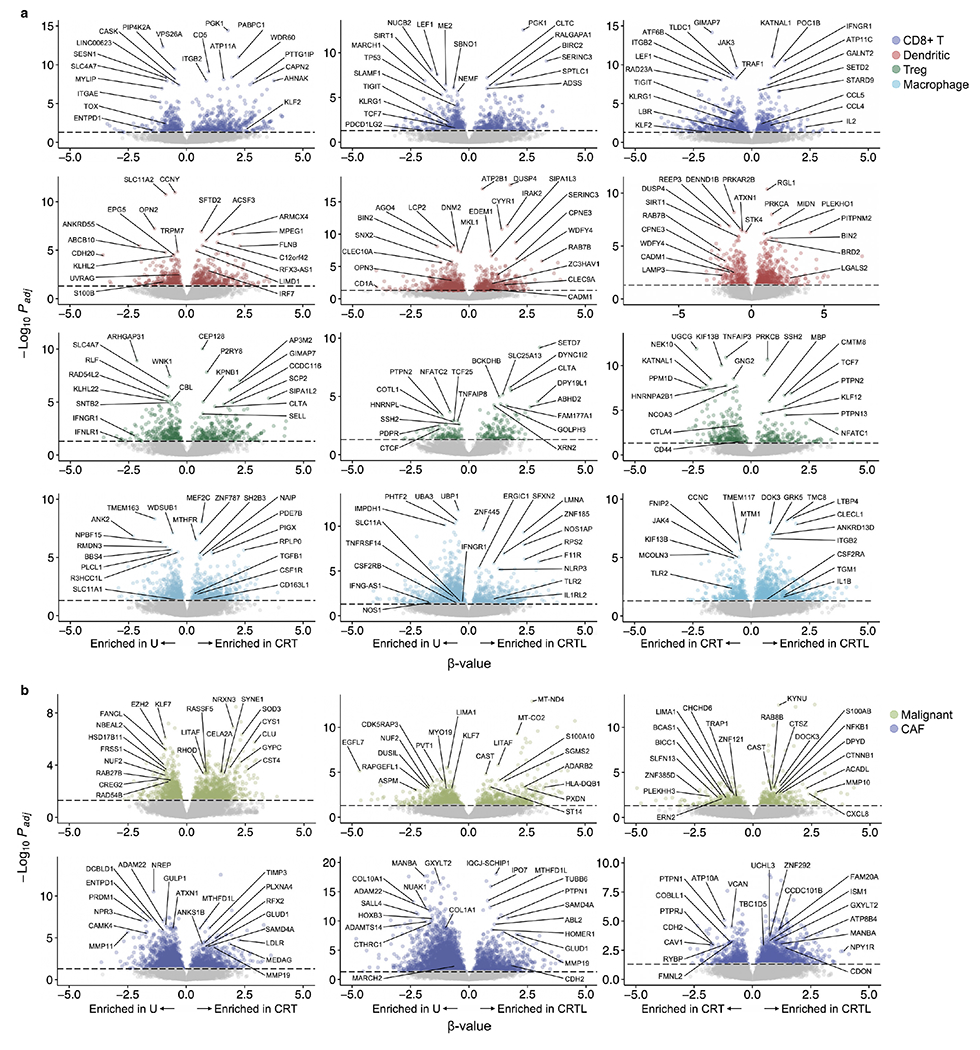
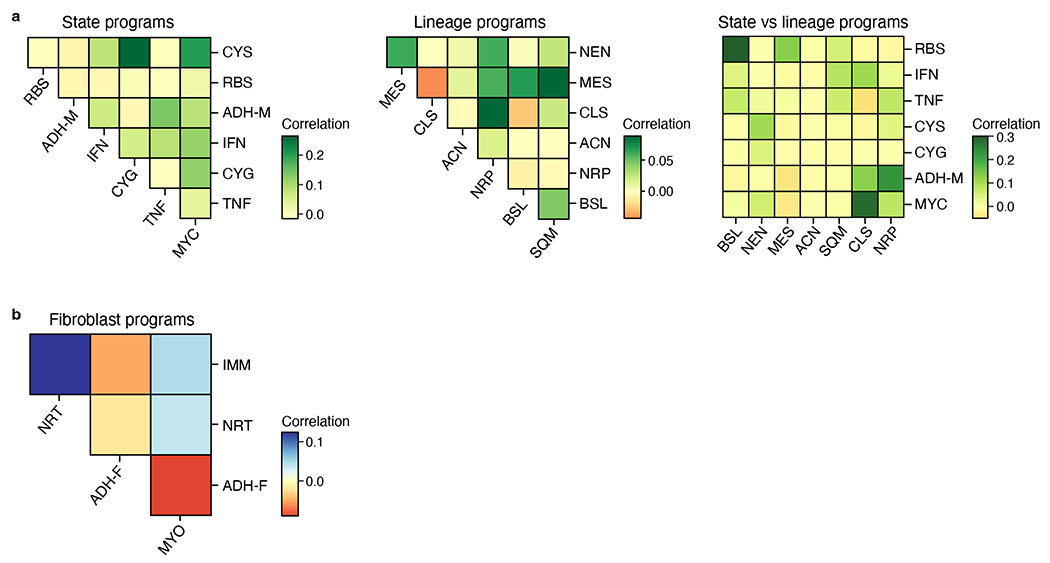
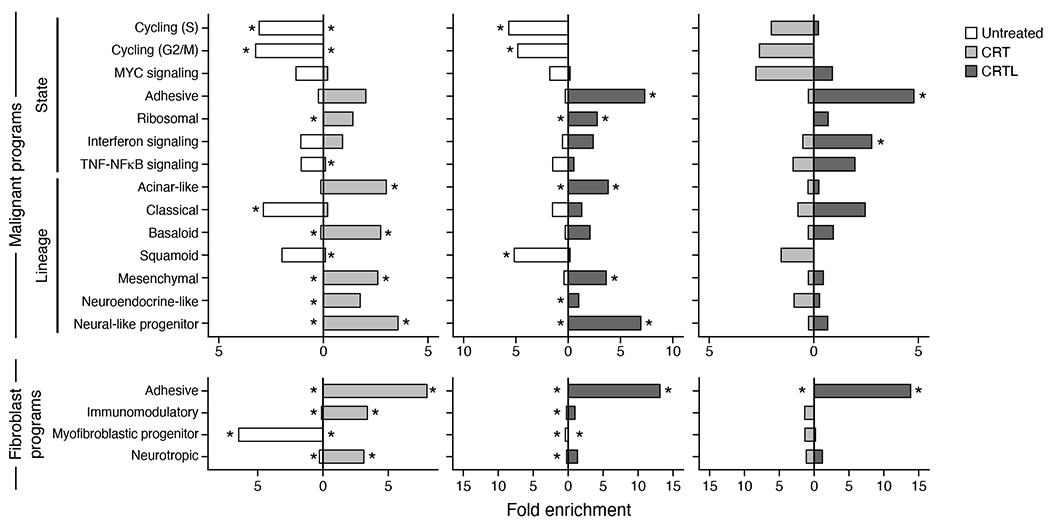
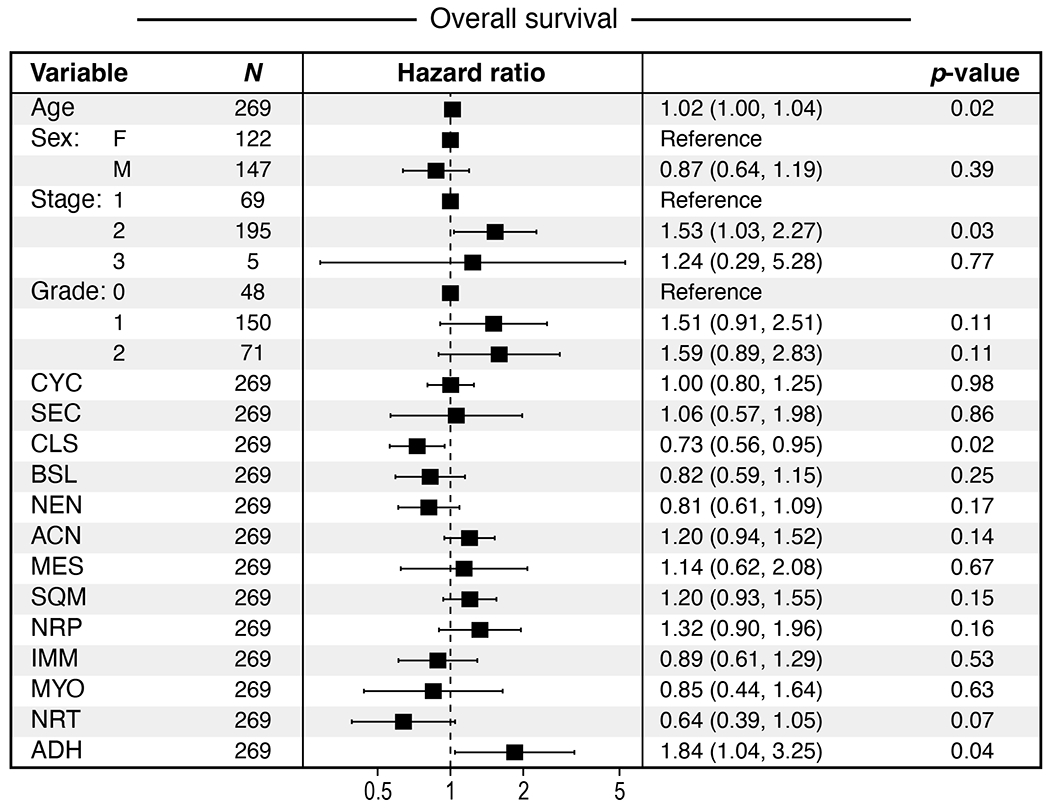
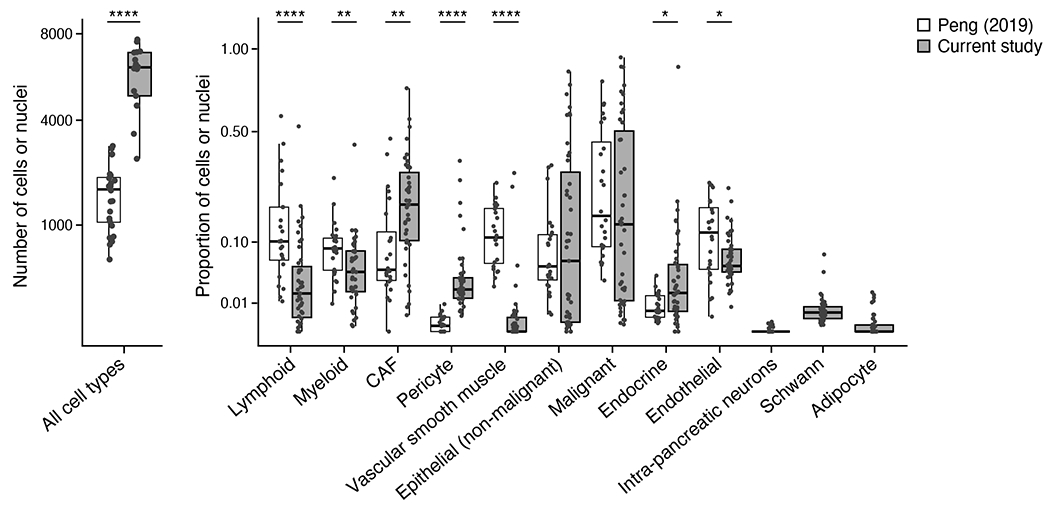
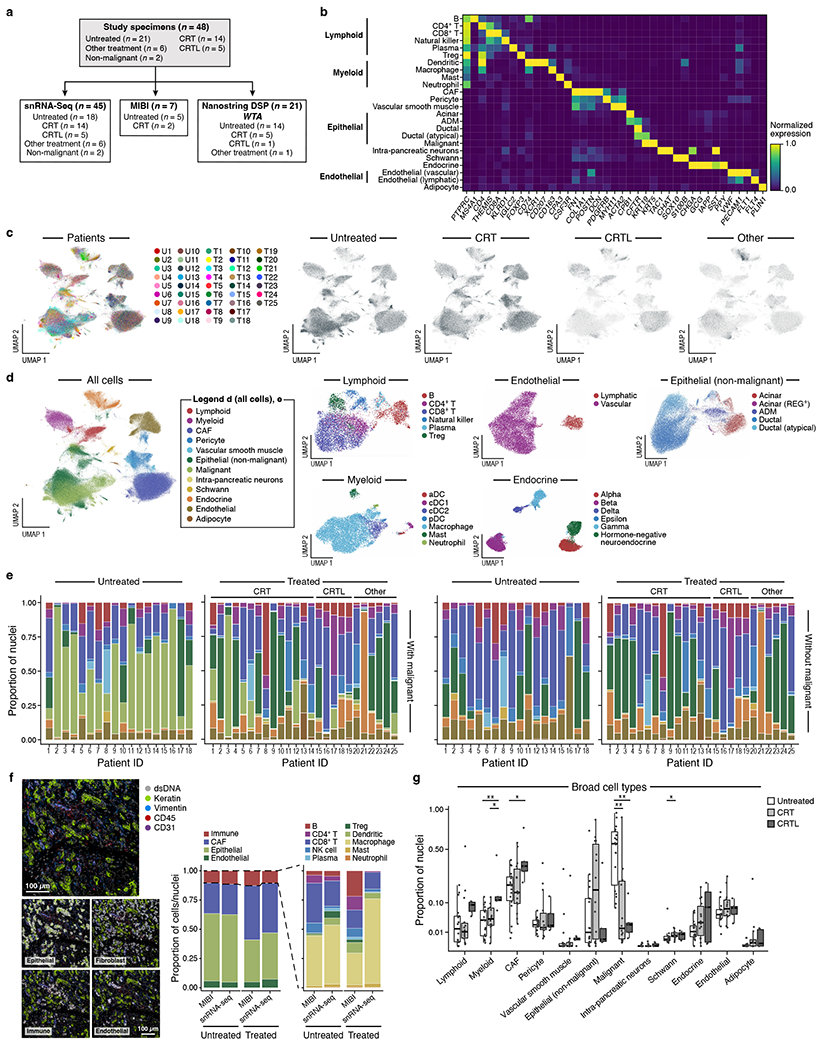
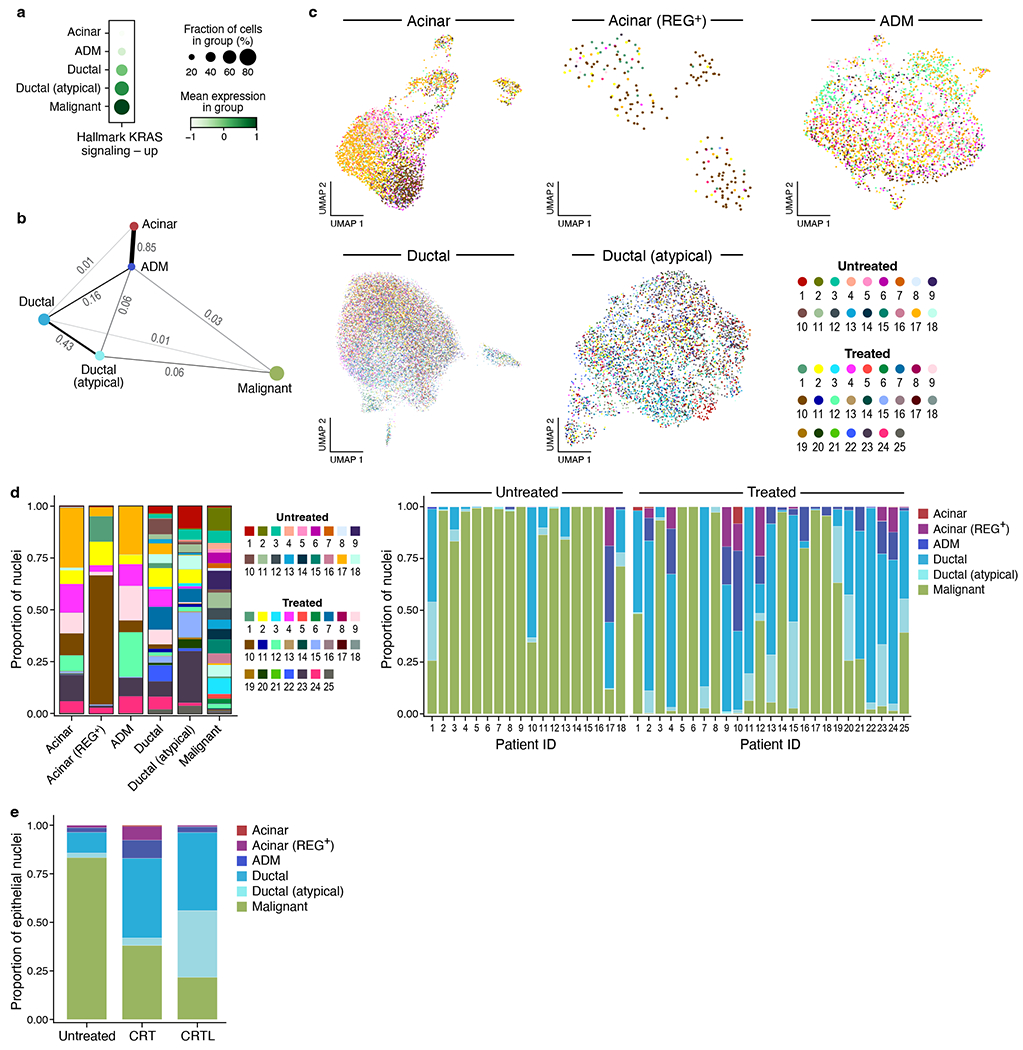
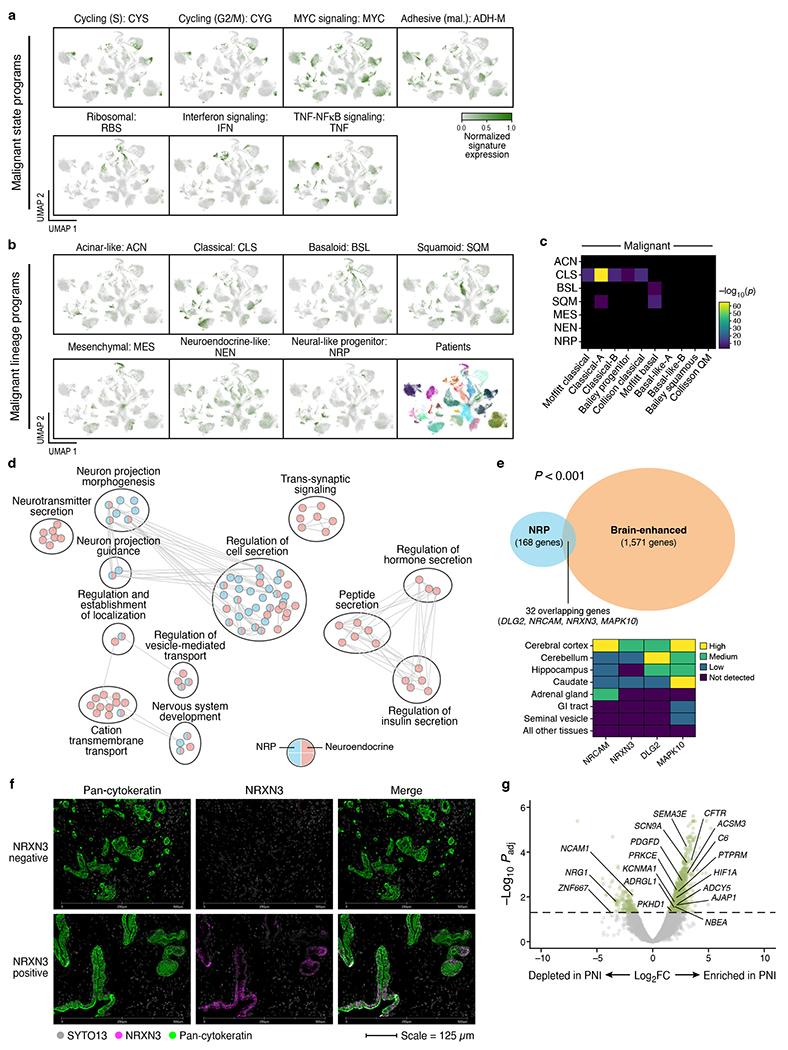
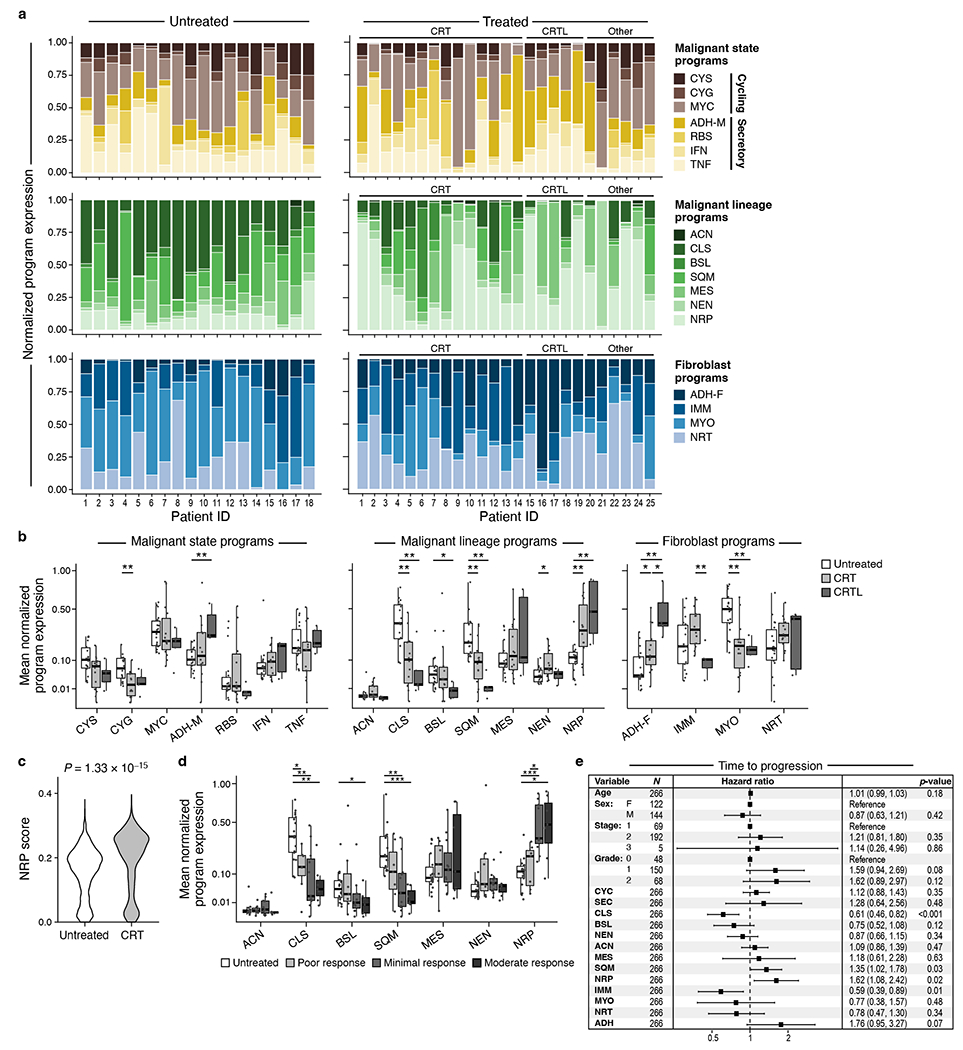
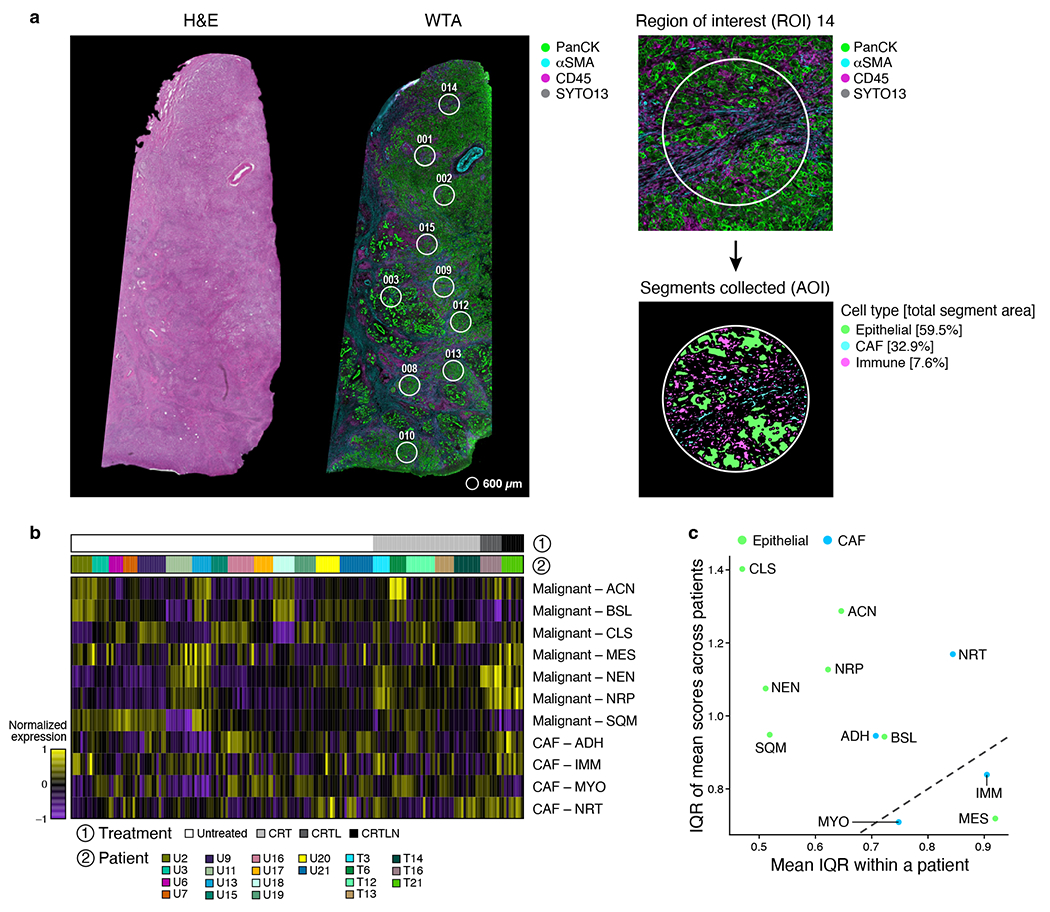
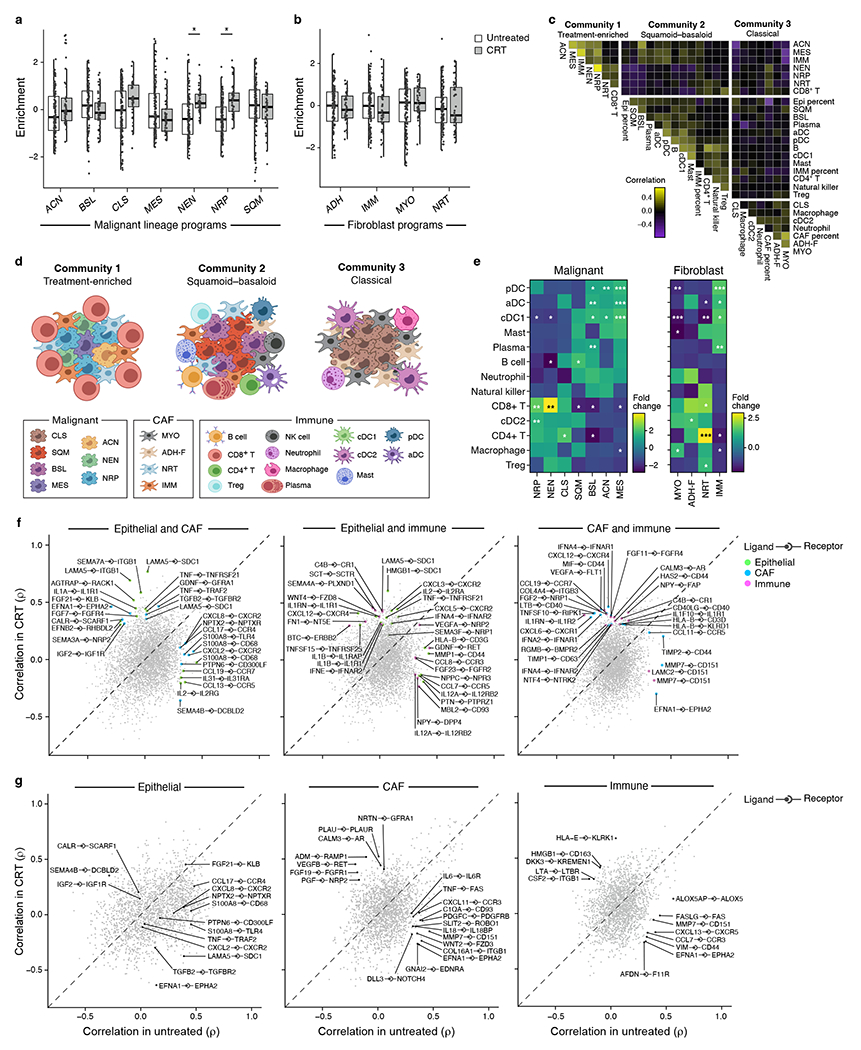
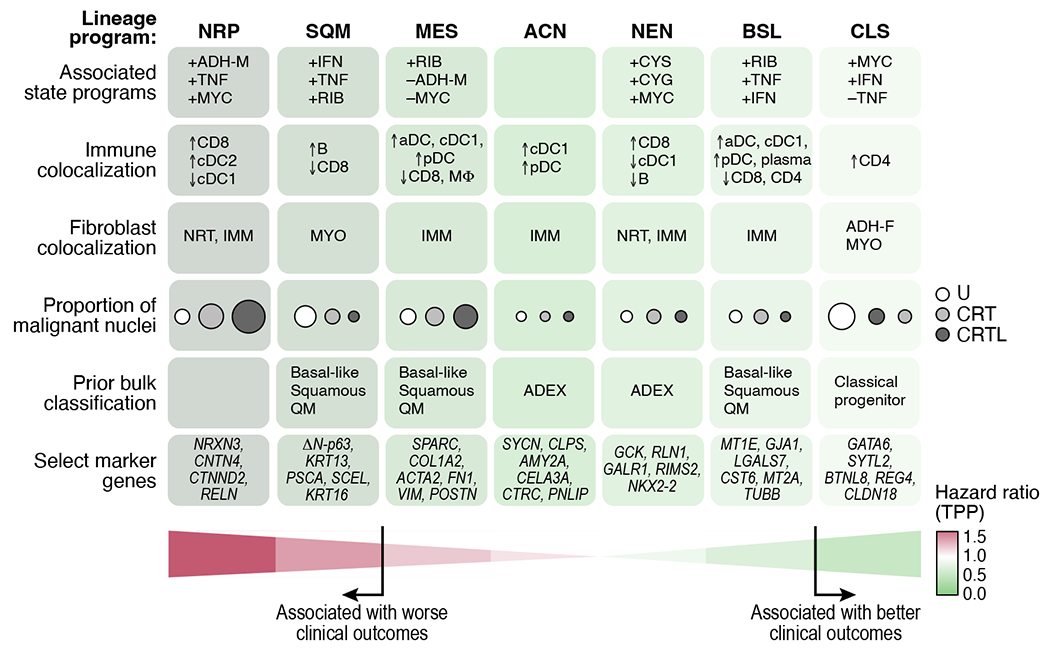
Pancreatic ductal adenocarcinoma (PDAC) is a highly lethal and treatment-refractory cancer. Molecular stratification in pancreatic cancer remains rudimentary and does not yet inform clinical management or therapeutic development. Here, we construct a high-resolution molecular landscape of the cellular subtypes and spatial communities that compose PDAC using single-nucleus RNA sequencing and whole-transcriptome digital spatial profiling (DSP) of 43 primary PDAC tumor specimens that either received neoadjuvant therapy or were treatment naive. We uncovered recurrent expression programs across malignant cells and fibroblasts, including a newly identified neural-like progenitor malignant cell program that was enriched after chemotherapy and radiotherapy and associated with poor prognosis in independent cohorts. Integrating spatial and cellular profiles revealed three multicellular communities with distinct contributions from malignant, fibroblast and immune subtypes: classical, squamoid-basaloid and treatment enriched. Our refined molecular and cellular taxonomy can provide a framework for stratification in clinical trials and serve as a roadmap for therapeutic targeting of specific cellular phenotypes and multicellular interactions.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Conflict of interest statement
A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov. From August 1, 2020, A.R. is an employee of Genentech. T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, and a co-Founder of Dragonfly Therapeutics and T2 Biosystems. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. Dr. Jacks’ laboratory also currently receives funding from Johnson & Johnson, but this fund did not support the research described in this manuscript. D.T.T. has received consulting fees from Nanostring Technologies, which was used in this work. D.T.T. has received consulting fees from ROME Therapeutics, Foundation Medicine, Inc., EMD Millipore Sigma, and Pfizer that are not related to this work. D.T.T. is a founder and has equity in ROME Therapeutics, PanTher Therapeutics and TellBio, Inc., which is not related to this work. D.T.T. receives research support from ACD-Biotechne, PureTech Health LLC, Ribon Therapeutics, which was not used in this work. M.M.K. has served as a compensated consultant for H3 Biomedicine and AstraZeneca and received a research grant (to institution) from Novartis that is not related to this work. R.K.J. received consultant fees from Elpis, Pfizer, SPARC, SynDevRx; owns equity in Accurius, Enlight, SynDevRx; serves on the Board of Trustees of Tekla Healthcare Investors, Tekla Life Sciences Investors, Tekla World Healthcare Fund and received a Research Grant from Boehringer Ingelheim all not related to this work. The interests of D.T.T., M.M.K., and R.K.J. were reviewed and are managed by Massachusetts General Hospital and Mass General Brigham in accordance with their conflict of interest policies. O.R.R. is a co-inventor on patent applications filed by the Broad Institute for inventions related to single cell genomics. ORR is an employee of Genentech since October 19, 2020. W.L.H., K.A.J., J.A.G., H.I.H., T.J., and A.R. are co-inventors on U.S. Provisional Patent Application No. 63/313,596 related to this work. All other authors declare no competing interests.
Figures






References
Extended Data References
-
- Bailey P et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 531, 47–52 (2016). - PubMed
-
- Collisson EA, Bailey P, Chang DK & Biankin AV Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol 16, 207–220 (2019). - PubMed
-
- Chan-Seng-Yue M et al. Transcription phenotypes of pancreatic cancer are driven by genomic events during tumor evolution. Nat. Genet 52, 231–240 (2020). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
