

Genome Sequence Variations of Infectious Bronchitis Virus Serotypes From Commercial Chickens in Mexico
- PMID: 35903135
- PMCID: PMC9315362
- DOI: 10.3389/fvets.2022.931272
Genome Sequence Variations of Infectious Bronchitis Virus Serotypes From Commercial Chickens in Mexico
Abstract
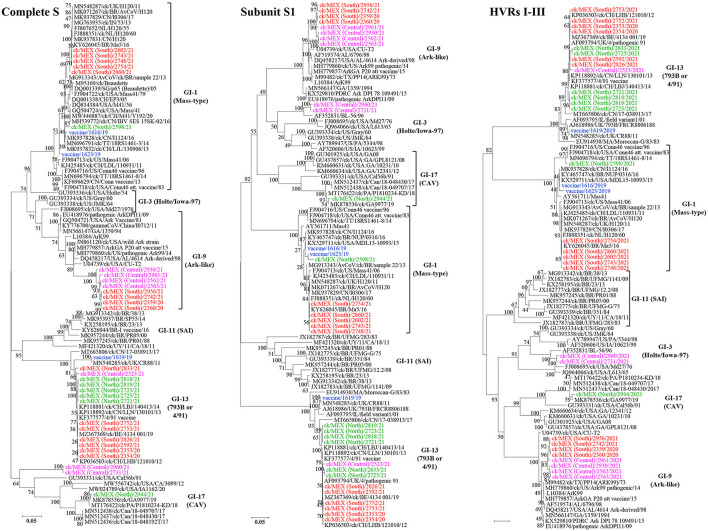
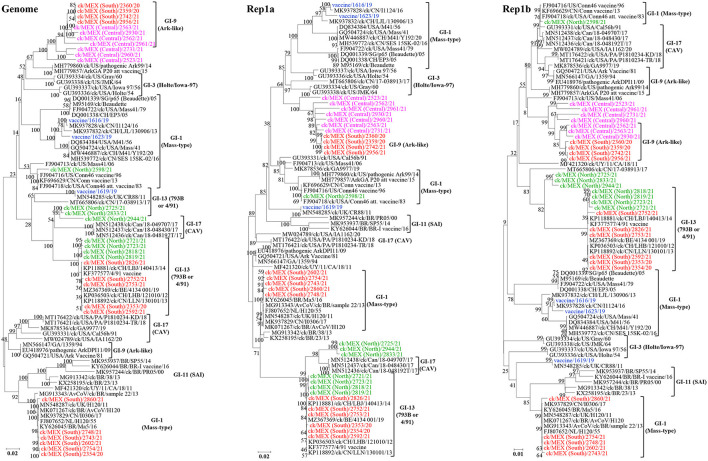
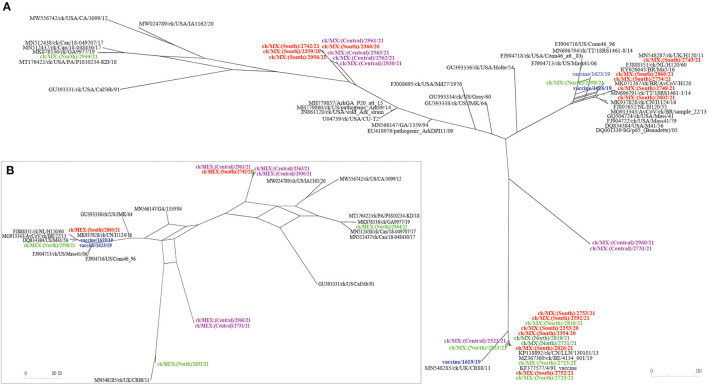
New variants of infectious bronchitis viruses (IBVs; Coronaviridae) continuously emerge despite routine vaccinations. Here, we report genome sequence variations of IBVs identified by random non-targeted next generation sequencing (NGS) of vaccine and field samples collected on FTA cards from commercial flocks in Mexico in 2019-2021. Paired-ended sequencing libraries prepared from rRNA-depleted RNAs were sequenced using Illumina MiSeq. IBV RNA was detected in 60.07% (n = 167) of the analyzed samples, from which 33 complete genome sequences were de novo assembled. The genomes are organized as 5'UTR-[Rep1a-Rep1b-S-3a-3b-E-M-4b-4c-5a-5b-N-6b]-3'UTR, except in eight sequences lacking non-structural protein genes (accessory genes) 4b, 4c, and 6b. Seventeen sequences have auxiliary S2' cleavage site located 153 residues downstream the canonically conserved primary furin-specific S1/S2 cleavage site. The sequences distinctly cluster into lineages GI-1 (Mass-type; n = 8), GI-3 (Holte/Iowa-97; n = 2), GI-9 (Arkansas-like; n = 8), GI-13 (793B; n = 14), and GI-17 (California variant; CAV; n = 1), with regional distribution in Mexico; this is the first report of the presence of 793B- and CAV-like strains in the country. Various point mutations, substitutions, insertions and deletions are present in the S1 hypervariable regions (HVRs I-III) across all 5 lineages, including in residues 38, 43, 56, 63, 66, and 69 that are critical in viral attachment to respiratory tract tissues. Nine intra-/inter-lineage recombination events are present in the S proteins of three Mass-type sequences, two each of Holte/Iowa-97 and Ark-like sequence, and one each of 793B-like and CAV-like sequences. This study demonstrates the feasibility of FTA cards as an attractive, adoptable low-cost sampling option for untargeted discovery of avian viral agents in field-collected clinical samples. Collectively, our data points to co-circulation of multiple distinct IBVs in Mexican commercial flocks, underscoring the need for active surveillance and a review of IBV vaccines currently used in Mexico and the larger Latin America region.
Keywords: NGS; hypervariable region; lineage; mutation; recombination; vaccine.
Copyright © 2022 Kariithi, Volkening, Leyson, Afonso, Christy, Decanini, Lemiere and Suarez.
Conflict of interest statement
JV and CA were employed by BASE2BIO. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


Similar articles
-
Molecular Characterization of Complete Genome Sequence of an Avian Coronavirus Identified in a Backyard Chicken from Tanzania.Genes (Basel). 2023 Sep 23;14(10):1852. doi: 10.3390/genes14101852. Genes (Basel). 2023. PMID: 37895200 Free PMC article.
-
Unique Variants of Avian Coronaviruses from Indigenous Chickens in Kenya.Viruses. 2023 Jan 17;15(2):264. doi: 10.3390/v15020264. Viruses. 2023. PMID: 36851482 Free PMC article.
-
Molecular epidemiology of infectious bronchitis virus in Poland from 1980 to 2017.Infect Genet Evol. 2020 Jun;80:104177. doi: 10.1016/j.meegid.2020.104177. Epub 2020 Jan 7. Infect Genet Evol. 2020. PMID: 31917362 Free PMC article.
-
Emergence and Dissemination of the Avian Infectious Bronchitis Virus Lineages in Poultry Farms in South America.Vet Sci. 2025 May 2;12(5):435. doi: 10.3390/vetsci12050435. Vet Sci. 2025. PMID: 40431528 Free PMC article. Review.
-
Current situation, genetic relationship and control measures of infectious bronchitis virus variants circulating in African regions.J Basic Appl Zool. 2016 Aug;76:20-30. doi: 10.1016/j.jobaz.2016.08.002. Epub 2016 Nov 20. J Basic Appl Zool. 2016. PMID: 32288944 Free PMC article. Review.
Cited by
-
Molecular Characterization of Complete Genome Sequence of an Avian Coronavirus Identified in a Backyard Chicken from Tanzania.Genes (Basel). 2023 Sep 23;14(10):1852. doi: 10.3390/genes14101852. Genes (Basel). 2023. PMID: 37895200 Free PMC article.
-
Genome Variability of Infectious Bronchitis Virus in Mexico: High Lineage Diversity and Recurrent Recombination.Viruses. 2023 Jul 20;15(7):1581. doi: 10.3390/v15071581. Viruses. 2023. PMID: 37515267 Free PMC article.
-
Virulent Newcastle disease virus genotypes V.3, VII.2, and XIII.1.1 and their coinfections with infectious bronchitis viruses and other avian pathogens in backyard chickens in Tanzania.Front Vet Sci. 2023 Oct 19;10:1272402. doi: 10.3389/fvets.2023.1272402. eCollection 2023. Front Vet Sci. 2023. PMID: 37929287 Free PMC article.
-
Key Aspects of Coronavirus Avian Infectious Bronchitis Virus.Pathogens. 2023 May 11;12(5):698. doi: 10.3390/pathogens12050698. Pathogens. 2023. PMID: 37242368 Free PMC article. Review.
-
Coding-complete genome sequence of a GI-13 infectious bronchitis virus from commercial chicken in India.Microbiol Resour Announc. 2025 Mar 11;14(3):e0114224. doi: 10.1128/mra.01142-24. Epub 2025 Jan 27. Microbiol Resour Announc. 2025. PMID: 39868773 Free PMC article.
References
-
- Jackwood MW, de Wit S. CHAPTER 4: Infectious Bronchitis. In: Swayne DE, Boulianne CM, Logue CM, McDougald LR, Nair V, Suarez DL, de Wit S, Grimes T, Johnson D, Kromm M, et al.., editors. Diseases of Poultry. Hoboken, NJ: John Wiley & Sons, Ltd. (2020). p. 167–88.
LinkOut - more resources
Full Text Sources
Research Materials