

Transgressive Potential Prediction and Optimal Cross Design of Seed Protein Content in the Northeast China Soybean Population Based on Full Exploration of the QTL-Allele System
- PMID: 35903228
- PMCID: PMC9317943
- DOI: 10.3389/fpls.2022.896549
Transgressive Potential Prediction and Optimal Cross Design of Seed Protein Content in the Northeast China Soybean Population Based on Full Exploration of the QTL-Allele System
Abstract
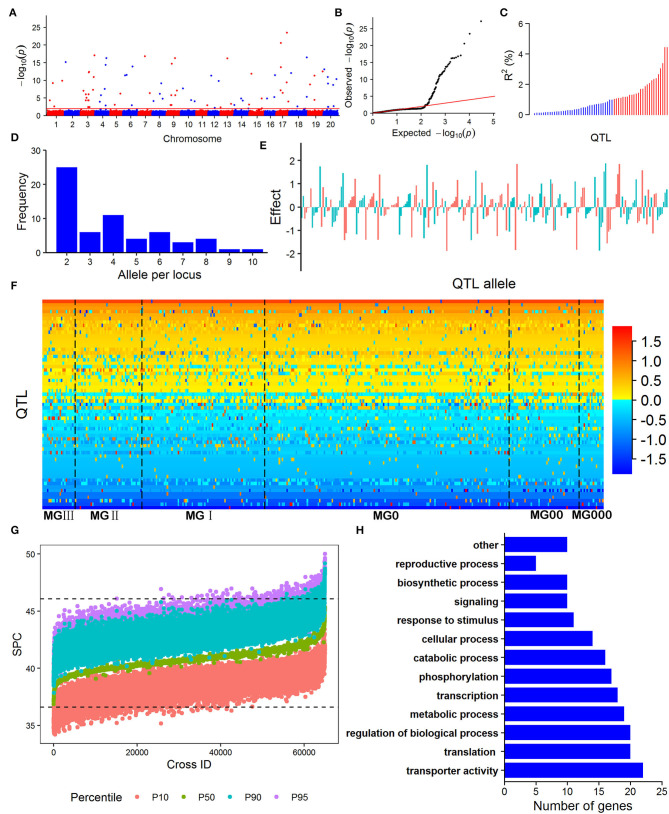
Northeast China is a major soybean production region in China. A representative sample of the Northeast China soybean germplasm population (NECSGP) composed of 361 accessions was evaluated for their seed protein content (SPC) in Tieling, Northeast China. This SPC varied greatly, with a mean SPC of 40.77%, ranging from 36.60 to 46.07%, but it was lower than that of the Chinese soybean landrace population (43.10%, ranging from 37.51 to 50.46%). The SPC increased slightly from 40.32-40.97% in the old maturity groups (MG, MGIII + II + I) to 40.93-41.58% in the new MGs (MG0 + 00 + 000). The restricted two-stage multi-locus genome-wide association study (RTM-GWAS) with 15,501 SNP linkage-disequilibrium block (SNPLDB) markers identified 73 SPC quantitative trait loci (QTLs) with 273 alleles, explaining 71.70% of the phenotypic variation, wherein 28 QTLs were new ones. The evolutionary changes of QTL-allele structures from old MGs to new MGs were analyzed, and 97.79% of the alleles in new MGs were inherited from the old MGs and 2.21% were new. The small amount of new positive allele emergence and possible recombination between alleles might explain the slight SPC increase in the new MGs. The prediction of recombination potentials in the SPC of all the possible crosses indicated that the mean of SPC overall crosses was 43.29% (+2.52%) and the maximum was 50.00% (+9.23%) in the SPC, and the maximum transgressive potential was 3.93%, suggesting that SPC breeding potentials do exist in the NECSGP. A total of 120 candidate genes were annotated and functionally classified into 13 categories, indicating that SPC is a complex trait conferred by a gene network.
Keywords: Northeast China soybean germplasm population (NECSGP); QTL-allele matrix; optimal cross prediction; restricted two stage multi-locus model GWAS (RTM-GWAS); seed protein content (SPC); transgressive potential.
Copyright © 2022 Feng, Fu, Fu, Sang, Wang, Wang, Ren, Du, Hao, Sun, Zhang, Wang, Xing, He and Gai.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Gene-allele system of shade tolerance in southern China soybean germplasm revealed by genome-wide association study using gene-allele sequence as markers.Theor Appl Genet. 2023 Jun 13;136(7):152. doi: 10.1007/s00122-023-04390-2. Theor Appl Genet. 2023. PMID: 37310498 Review.
-
Identification of QTL-allele systems of seed size and oil content for simultaneous genomic improvement in Northeast China soybeans.Front Plant Sci. 2024 Nov 14;15:1483995. doi: 10.3389/fpls.2024.1483995. eCollection 2024. Front Plant Sci. 2024. PMID: 39610887 Free PMC article.
-
Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes.J Exp Bot. 2015 Oct;66(20):6311-25. doi: 10.1093/jxb/erv342. Epub 2015 Jul 10. J Exp Bot. 2015. PMID: 26163701
-
Detecting the QTL-Allele System of Seed Oil Traits Using Multi-Locus Genome-Wide Association Analysis for Population Characterization and Optimal Cross Prediction in Soybean.Front Plant Sci. 2018 Dec 5;9:1793. doi: 10.3389/fpls.2018.01793. eCollection 2018. Front Plant Sci. 2018. PMID: 30568668 Free PMC article.
-
Detecting the QTL-allele system of seed isoflavone content in Chinese soybean landrace population for optimal cross design and gene system exploration.Theor Appl Genet. 2016 Aug;129(8):1557-76. doi: 10.1007/s00122-016-2724-0. Epub 2016 May 17. Theor Appl Genet. 2016. PMID: 27189002
Cited by
-
Gene-allele system of shade tolerance in southern China soybean germplasm revealed by genome-wide association study using gene-allele sequence as markers.Theor Appl Genet. 2023 Jun 13;136(7):152. doi: 10.1007/s00122-023-04390-2. Theor Appl Genet. 2023. PMID: 37310498 Review.
-
An Improved Genome-Wide Association Procedure Explores Gene-Allele Constitutions and Evolutionary Drives of Growth Period Traits in the Global Soybean Germplasm Population.Int J Mol Sci. 2023 May 31;24(11):9570. doi: 10.3390/ijms24119570. Int J Mol Sci. 2023. PMID: 37298521 Free PMC article.
References
LinkOut - more resources
Full Text Sources