

JAK2 Alterations in Acute Lymphoblastic Leukemia: Molecular Insights for Superior Precision Medicine Strategies
- PMID: 35903543
- PMCID: PMC9315936
- DOI: 10.3389/fcell.2022.942053
JAK2 Alterations in Acute Lymphoblastic Leukemia: Molecular Insights for Superior Precision Medicine Strategies
Abstract
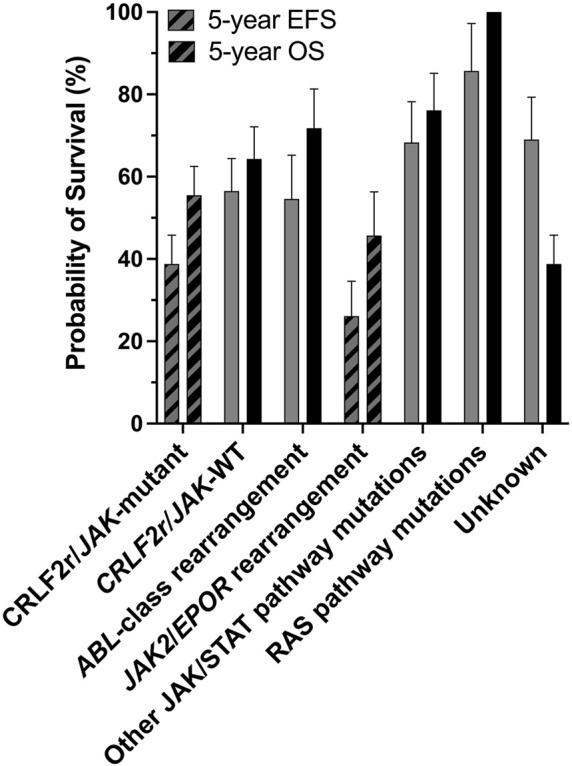
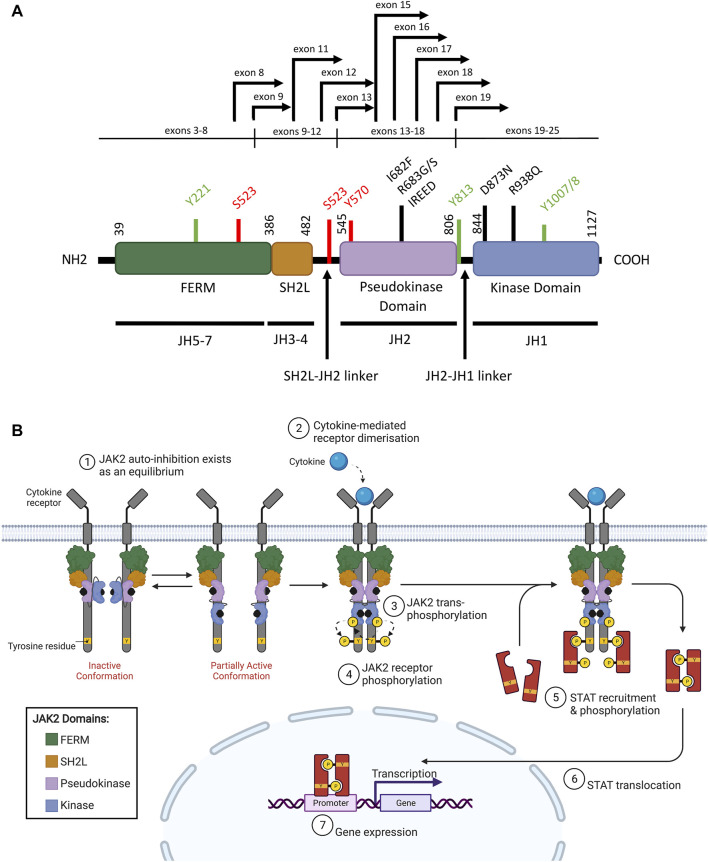
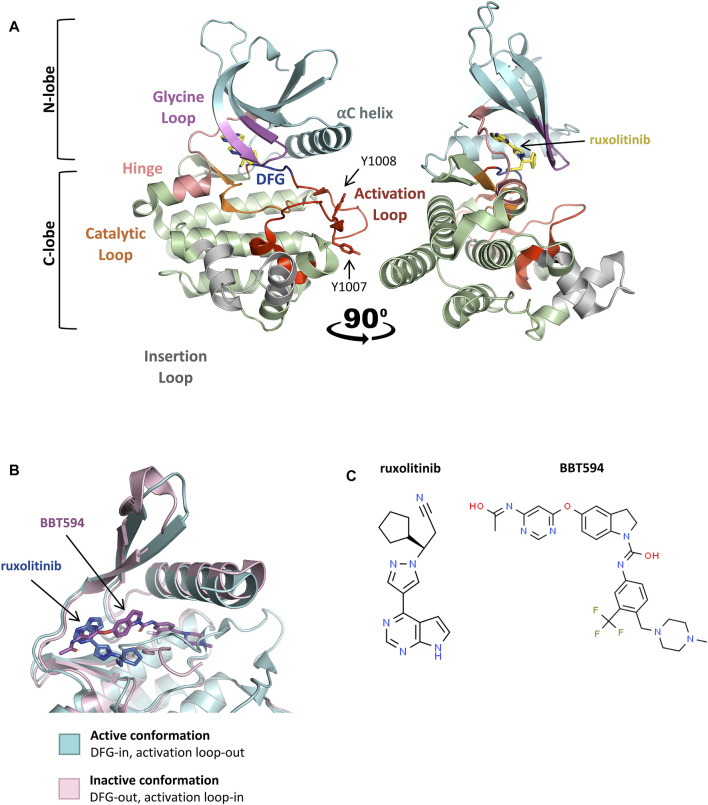
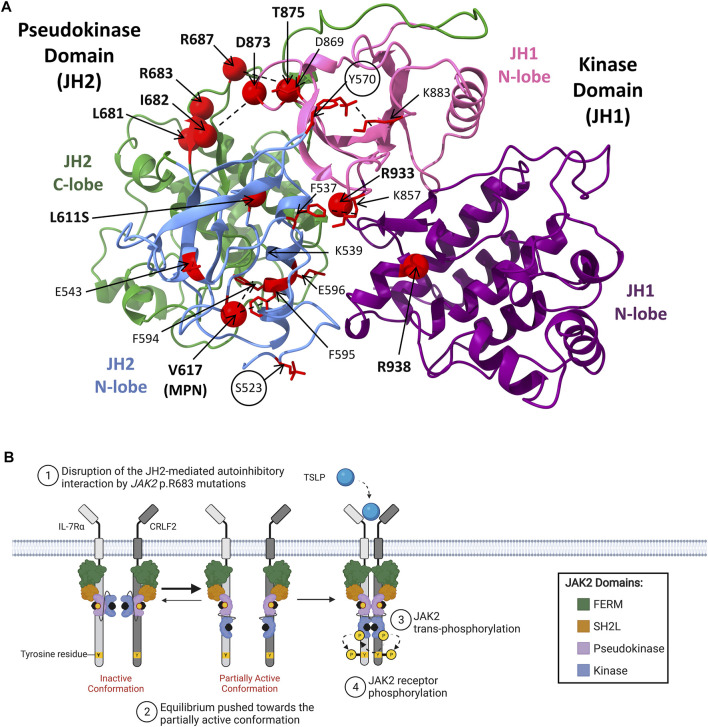
Acute lymphoblastic leukemia (ALL) is the most common pediatric cancer, arising from immature lymphocytes that show uncontrolled proliferation and arrested differentiation. Genomic alterations affecting Janus kinase 2 (JAK2) correlate with some of the poorest outcomes within the Philadelphia-like subtype of ALL. Given the success of kinase inhibitors in the treatment of chronic myeloid leukemia, the discovery of activating JAK2 point mutations and JAK2 fusion genes in ALL, was a breakthrough for potential targeted therapies. However, the molecular mechanisms by which these alterations activate JAK2 and promote downstream signaling is poorly understood. Furthermore, as clinical data regarding the limitations of approved JAK inhibitors in myeloproliferative disorders matures, there is a growing awareness of the need for alternative precision medicine approaches for specific JAK2 lesions. This review focuses on the molecular mechanisms behind ALL-associated JAK2 mutations and JAK2 fusion genes, known and potential causes of JAK-inhibitor resistance, and how JAK2 alterations could be targeted using alternative and novel rationally designed therapies to guide precision medicine approaches for these high-risk subtypes of ALL.
Keywords: JAK2; Janus kinases; acute lymphoblastic leukemia; kinase inhibitor; leukemia; targeted therapy.
Copyright © 2022 Downes, McClure, McDougal, Heatley, Bruning, Thomas, Yeung and White.
Conflict of interest statement
DW receives research support from BMS, Honoraria from Amgen. DY receives research support from Novartis, Ariad and BMS, Honoraria from Novartis, BMS, Amgen and Pfizer. None of these agencies have had a role in the preparation of this manuscript. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
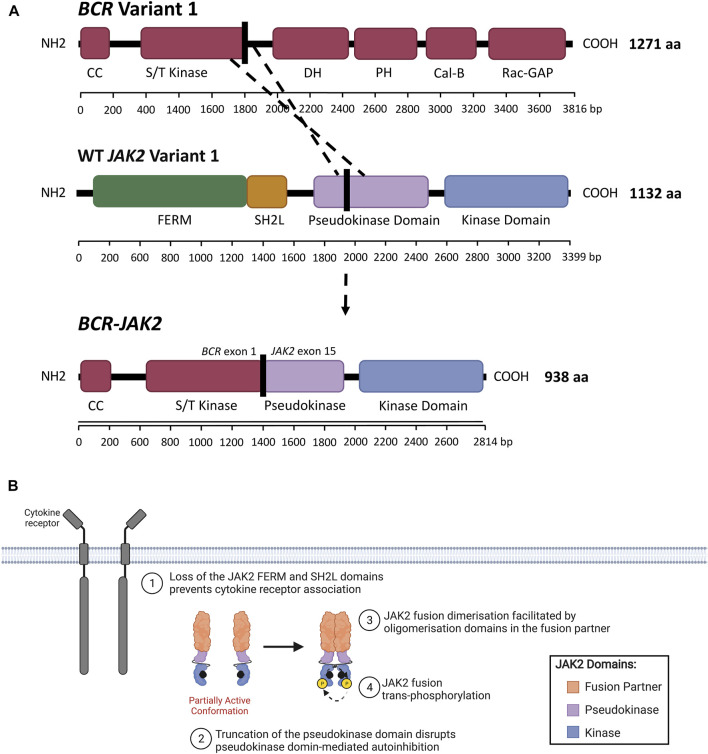
References
-
- Andraos R., Qian Z., Bonenfant D., Rubert J., Vangrevelinghe E., Scheufler C., et al. (2012). Modulation of Activation-Loop Phosphorylation by JAK Inhibitors Is Binding Mode Dependent. Cancer Discov. 2, 512–523. 10.1158/2159-8290.cd-11-0324 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous