

A retrospective analysis of endocrine disease in sphingosine-1-phosphate lyase insufficiency: case series and literature review
- PMID: 35904228
- PMCID: PMC9346324
- DOI: 10.1530/EC-22-0250
A retrospective analysis of endocrine disease in sphingosine-1-phosphate lyase insufficiency: case series and literature review
Abstract
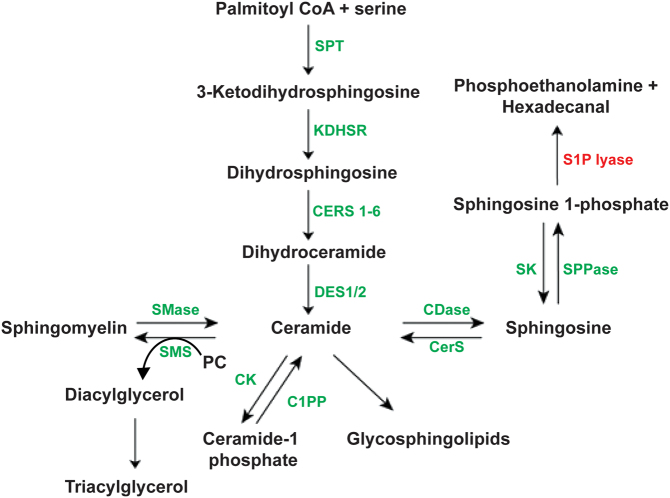
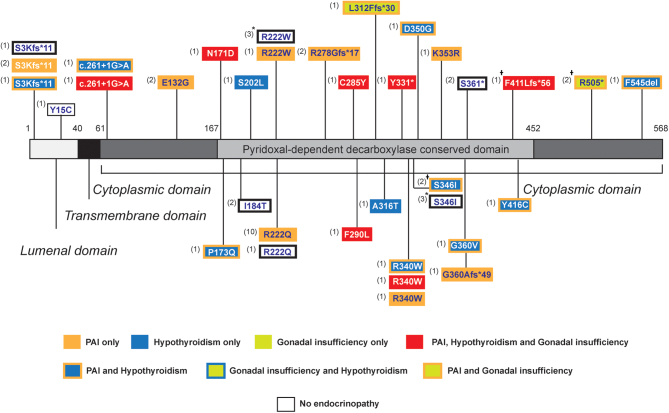
Sphingosine-1-phosphate lyase (SGPL1) insufficiency syndrome (SPLIS) is an autosomal recessive multi-system disorder, which mainly incorporates steroid-resistant nephrotic syndrome and primary adrenal insufficiency. Other variable endocrine manifestations are described. In this study, we aimed to comprehensively annotate the endocrinopathies associated with pathogenic SGPL1 variants and assess for genotype-phenotype correlations by retrospectively reviewing the reports of endocrine disease within our patient cohort and all published cases in the wider literature up to February 2022. Glucocorticoid insufficiency in early childhood is the most common endocrine manifestation affecting 64% of the 50 patients reported with SPLIS, and a third of these individuals have additional mineralocorticoid deficiency. While most individuals also have nephrotic syndrome, SGPL1 variants also account for isolated adrenal insufficiency at presentation. Primary gonadal insufficiency, manifesting with microphallus and cryptorchidism, is reported in less than one-third of affected boys, all with concomitant adrenal disease. Mild primary hypothyroidism affects approximately a third of patients. There is paucity of data on the impact of SGPL1 deficiency on growth, and pubertal development, limited by the early and high mortality rate (approximately 50%). There is no clear genotype-phenotype correlation overall in the syndrome, with variable disease penetrance within individual kindreds. However, with regards to endocrine phenotype, the most prevalent disease variant p.R222Q (affecting 22%) is most consistently associated with isolated glucocorticoid deficiency. To conclude, SPLIS is associated with significant multiple endocrine disorders. While endocrinopathy in the syndrome generally presents in infancy, late-onset disease also occurs. Screening for these is therefore warranted both at diagnosis and through follow-up.
Keywords: SGPL1; primary adrenal insufficiency; primary gonadal insufficiency; primary hypothyroidism; sphingolipids; sphingosine-1-phosphate lyase.
Figures


Similar articles
-
Clinical presentation and management challenges of sphingosine-1-phosphate lyase insufficiency syndrome associated with an SGPL1 variant: a case report.BMC Pediatr. 2025 Jan 4;25(1):1. doi: 10.1186/s12887-024-05311-y. BMC Pediatr. 2025. PMID: 39755650 Free PMC article.
-
Sphingosine Phosphate Lyase Insufficiency Syndrome.2020 Oct 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2020 Oct 15. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 33074640 Free Books & Documents. Review.
-
Congenital adrenal calcifications as the first clinical indication of sphingosine lyase insufficiency syndrome: A case report and review of the literature.Am J Med Genet A. 2022 Nov;188(11):3312-3317. doi: 10.1002/ajmg.a.62956. Epub 2022 Aug 16. Am J Med Genet A. 2022. PMID: 35972040 Free PMC article. Review.
-
A Sphingosine-1-Phosphate Lyase Mutation Associated With Congenital Nephrotic Syndrome and Multiple Endocrinopathy.Front Pediatr. 2020 Apr 8;8:151. doi: 10.3389/fped.2020.00151. eCollection 2020. Front Pediatr. 2020. PMID: 32322566 Free PMC article.
-
Insights From Long-term Follow-up of a Girl With Adrenal Insufficiency and Sphingosine-1-Phosphate Lyase Deficiency.J Endocr Soc. 2022 Feb 11;6(5):bvac020. doi: 10.1210/jendso/bvac020. eCollection 2022 May 1. J Endocr Soc. 2022. PMID: 35308304 Free PMC article.
Cited by
-
Familial Glucocorticoid Deficiency: the changing landscape of an eponymous syndrome.Front Endocrinol (Lausanne). 2023 Dec 21;14:1268345. doi: 10.3389/fendo.2023.1268345. eCollection 2023. Front Endocrinol (Lausanne). 2023. PMID: 38189052 Free PMC article. Review.
-
Clinical presentation and management challenges of sphingosine-1-phosphate lyase insufficiency syndrome associated with an SGPL1 variant: a case report.BMC Pediatr. 2025 Jan 4;25(1):1. doi: 10.1186/s12887-024-05311-y. BMC Pediatr. 2025. PMID: 39755650 Free PMC article.
-
Addison's Disease: Diagnosis and Management Strategies.Int J Gen Med. 2023 Jun 2;16:2187-2210. doi: 10.2147/IJGM.S390793. eCollection 2023. Int J Gen Med. 2023. PMID: 37287503 Free PMC article. Review.
-
Combined novel homozygous variants in both SGPL1 and STAT1 presenting with severe combined immune deficiency: case report and literature review.Front Immunol. 2023 Jun 12;14:1186575. doi: 10.3389/fimmu.2023.1186575. eCollection 2023. Front Immunol. 2023. PMID: 37377976 Free PMC article. Review.
-
Adrenal insufficiency in inborn errors of metabolism and vice versa: Case reports and review of the literature.Mol Genet Metab Rep. 2025 May 26;43:101232. doi: 10.1016/j.ymgmr.2025.101232. eCollection 2025 Jun. Mol Genet Metab Rep. 2025. PMID: 40502453 Free PMC article. Review.
References
-
- Janecke AR, Xu R, Steichen-Gersdorf E, Waldegger S, Entenmann A, Giner T, Krainer I, Huber LA, Hess MW, Frishberg Yet al.Deficiency of the sphingosine-1-phosphate lyase SGPL1 is associated with congenital nephrotic syndrome and congenital adrenal calcifications. Human Mutation 201738365–372. (10.1002/humu.23192) - DOI - PMC - PubMed
-
- Prasad R, Hadjidemetriou I, Maharaj A, Meimaridou E, Buonocore F, Saleem M, Hurcombe J, Bierzynska A, Barbagelata E, Bergadá Iet al.Sphingosine-1-phosphate lyase mutations cause primary adrenal insufficiency and steroid-resistant nephrotic syndrome. Journal of Clinical Investigation 2017127942–953. (10.1172/JCI90171) - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
