

Impaired Proteolysis of Noncanonical RAS Proteins Drives Clonal Hematopoietic Transformation
- PMID: 35904492
- PMCID: PMC9533010
- DOI: 10.1158/2159-8290.CD-21-1631
Impaired Proteolysis of Noncanonical RAS Proteins Drives Clonal Hematopoietic Transformation
Abstract
Recently, screens for mediators of resistance to FLT3 and ABL kinase inhibitors in leukemia resulted in the discovery of LZTR1 as an adapter of a Cullin-3 RING E3 ubiquitin ligase complex responsible for the degradation of RAS GTPases. In parallel, dysregulated LZTR1 expression via aberrant splicing and mutations was identified in clonal hematopoietic conditions. Here we identify that loss of LZTR1, or leukemia-associated mutants in the LZTR1 substrate and RAS GTPase RIT1 that escape degradation, drives hematopoietic stem cell (HSC) expansion and leukemia in vivo. Although RIT1 stabilization was sufficient to drive hematopoietic transformation, transformation mediated by LZTR1 loss required MRAS. Proteolysis targeting chimeras (PROTAC) against RAS or reduction of GTP-loaded RAS overcomes LZTR1 loss-mediated resistance to FLT3 inhibitors. These data reveal proteolysis of noncanonical RAS proteins as novel regulators of HSC self-renewal, define the function of RIT1 and LZTR1 mutations in leukemia, and identify means to overcome drug resistance due to LZTR1 downregulation.
Significance: Here we identify that impairing proteolysis of the noncanonical RAS GTPases RIT1 and MRAS via LZTR1 downregulation or leukemia-associated mutations stabilizing RIT1 enhances MAP kinase activation and drives leukemogenesis. Reducing the abundance of GTP-bound KRAS and NRAS overcomes the resistance to FLT3 kinase inhibitors associated with LZTR1 downregulation in leukemia. This article is highlighted in the In This Issue feature, p. 2221.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
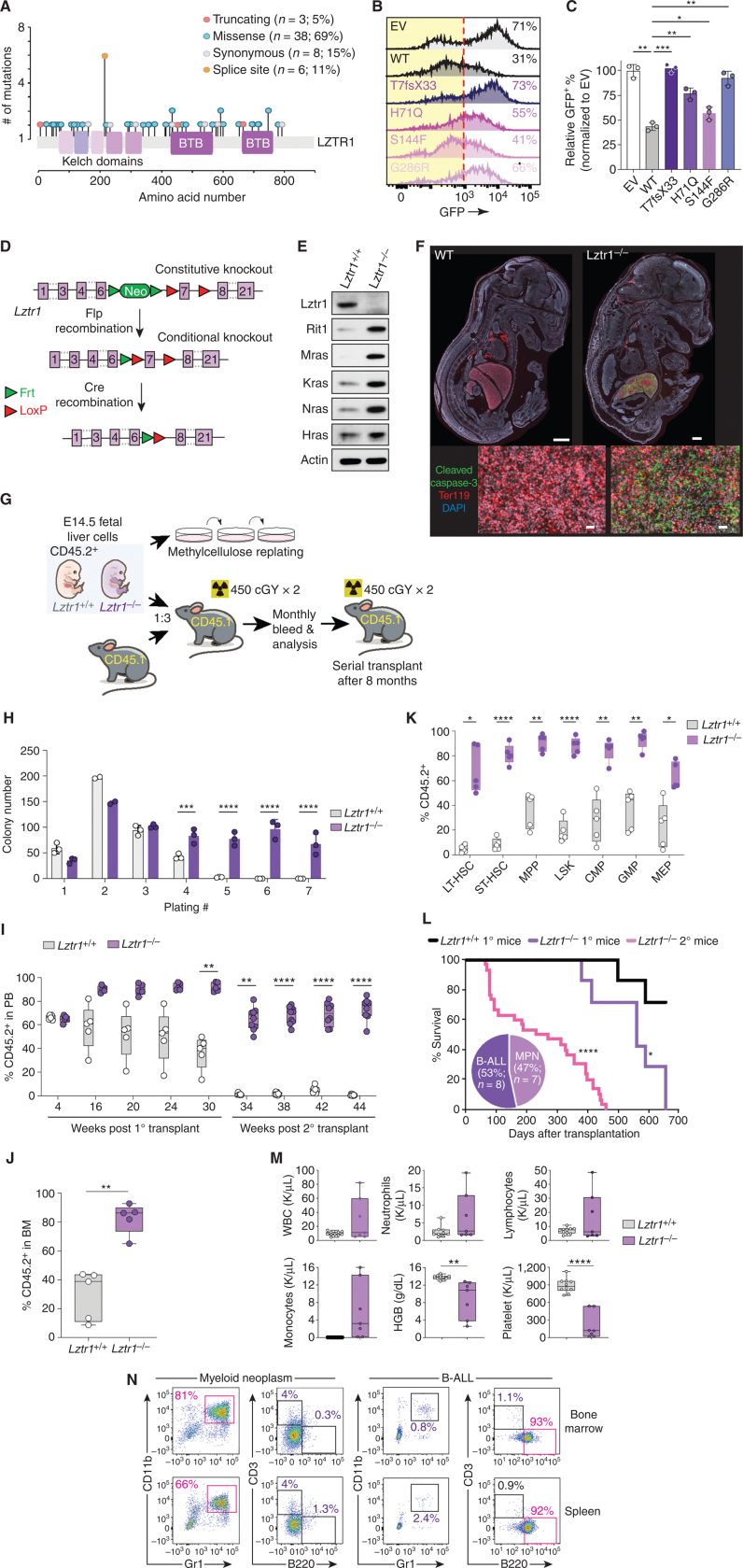
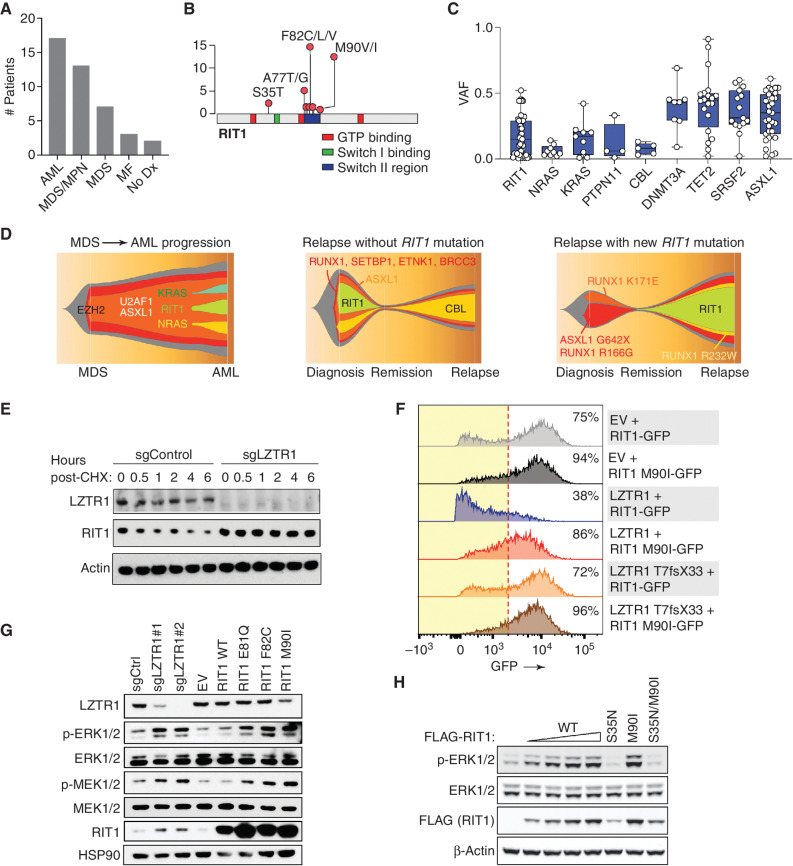
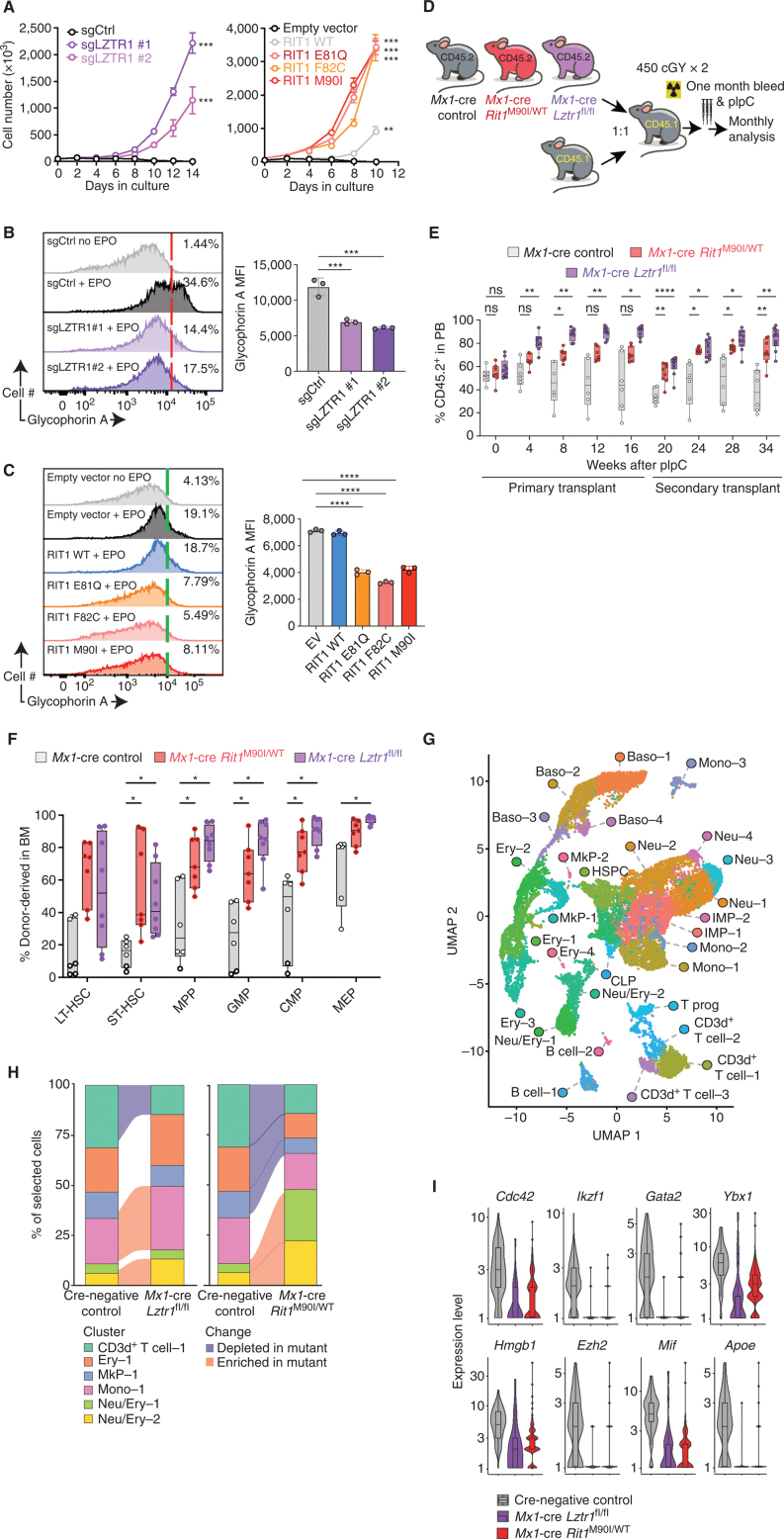
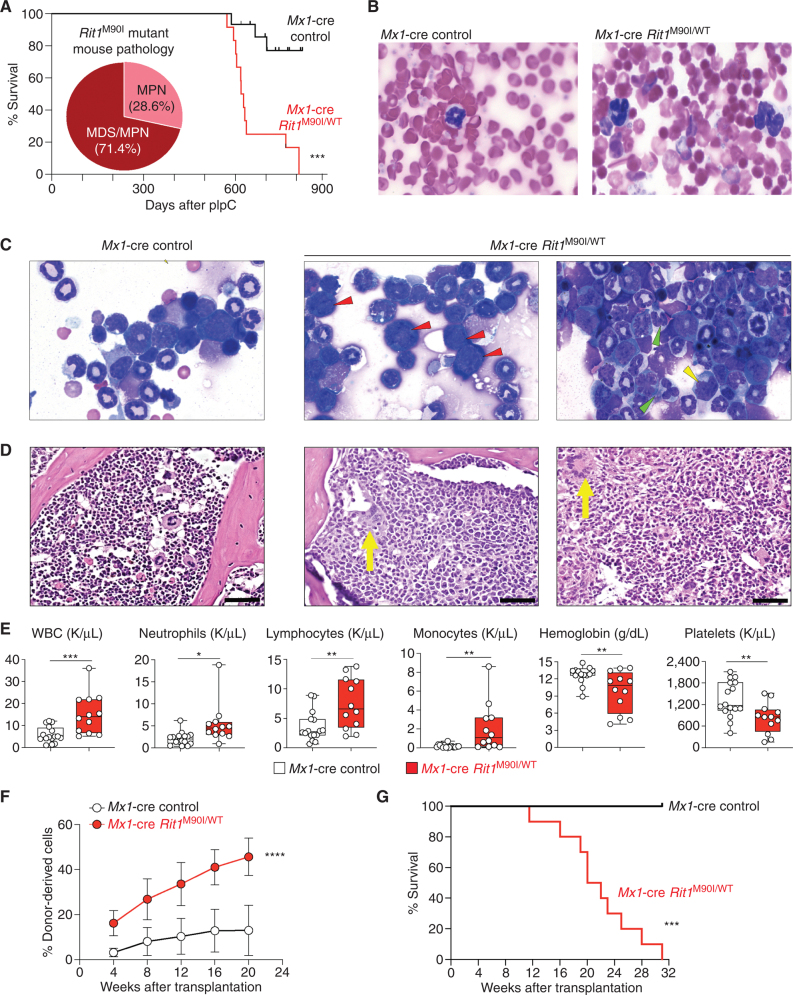
![Figure 6. LZTR1-mediated drug resistance can be overcome by modulating RAS-GTP abundance. A, Scatter plots of RNA expression levels [shown as normalized reads per kilobase of transcript per million reads mapped (RPKM)] plotted against drug-sensitivity measured ex vivo as the area under the curve (AUC) from AML patient samples harboring FLT3-ITD mutation. n ≥ 111. *, P < 0.05. Pearson correlation coefficients are shown as (r) values. Statistical significance is indicated. B, Schema of the MOLM-13 cell line xenograft experiment. Luciferase-expressing MOLM-13 cells treated with anti-LZTR1 or control sgRNAs were systemically engrafted with 1 × 105 cells/animal via a tail-vein injection after sublethal (225 cGy) irradiation. Seven days later, animals were treated with either vehicle or 30 mg/kg/day of gilteritinib via oral gavage. Animals underwent bioluminescence imaging weekly. C, Kaplan–Meier curve of experiment in B. n = 7–10. **, P < 0.01; ***, P < 0.001. D, Schema of protein-based degrader of RAS proteins. A chimeric protein consisting of a high-affinity target-binding domain for RAS proteins (DARPin K27) is fused to an engineered E3 ligase adapter (SPOP) to confer ubiquitin-mediated degradation of RAS proteins. E, Western blot demonstrating levels of LZTR1 as well as total and GTP-bound KRAS/NRAS/MRAS and RIT1 in MOLM-13 cells ± LZTR1 KO upon doxycycline-mediated induction of the RAS biodegrader (performed in biological triplicate). EV, empty vector. F, Seventy-two-hour cell proliferation assay on parental and LZTR1 KO MOLM-13 cells ± induction of RAS degradation. Cell viability was measured in triplicate using CellTiter-Glo. G, 2D synergy plots using the Zero Interaction Potency (ZIP) model of control sgRNA (“WT”) or anti-LZTR1 sgRNA (“LZTR1 KO”) MOLM-13 cells treated for 72 hours with BI-3406 and/or gilteritinib at various concentrations. Western blot demonstrating p-MEK, p-ERK, and total MEK, ERK, KRAS/NRAS/MRAS, and RIT1 levels (H) as well as RAS-GTP levels (I) in MOLM-13 cells ± LZTR1 KO treated with increasing concentration of DMSO or BI-3406 alone 4 hours after drug treatment.](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/a403/9533010/5b444dfc9322/2434fig6.jpg)
Comment in
- doi: 10.1158/2159-8290.CD-12-10-ITI
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
