

The role of amyloid β in the pathological mechanism of GNE myopathy
- PMID: 35904705
- PMCID: PMC9616754
- DOI: 10.1007/s10072-022-06301-7
The role of amyloid β in the pathological mechanism of GNE myopathy
Abstract
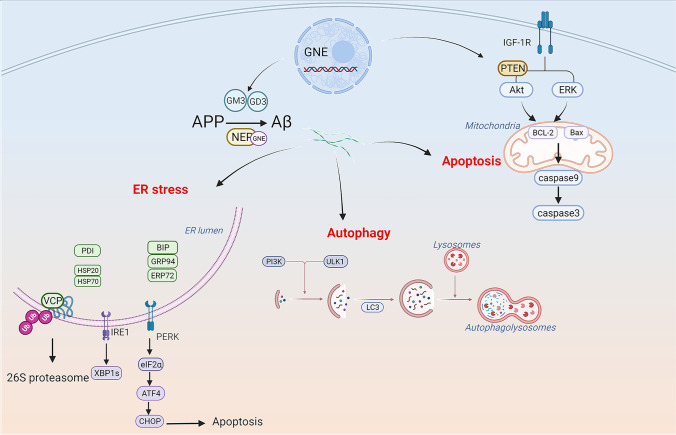
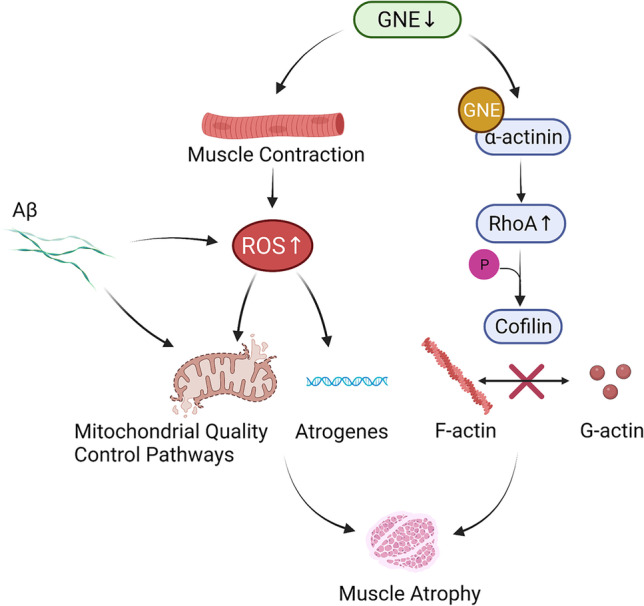
GNE myopathy is a hereditary muscle disorder characterized by muscle atrophy and weakness initially involving the lower distal extremities. The treatment of GNE myopathy mainly focuses on a sialic acid deficiency caused by a mutation in the GNE gene, but it has not achieved the expected effect. The main pathological features of GNE myopathy are myofiber atrophy and rimmed vacuoles, including accumulation of amyloid β, which is mainly found in atrophic muscle fibers. Although the role of amyloid β and other misfolded proteins on the nervous system has been widely recognized, the cause and process of the formation of amyloid β in the pathological process of GNE myopathy are unclear. In addition, amyloid β has been reported to be linked to quality control mechanisms of proteins, such as molecular chaperones, the ubiquitin-proteasome system, and the autophagy-lysosome system. Herein, we summarize the possible reasons for amyloid β deposition and illustrate amyloid β-mediated events in the cells and their role in muscle atrophy in GNE myopathy. This review represents an overview of amyloid β and GNE myopathy that could help identify a potential mechanism and thereby a plausible therapeutic for the disease.
Keywords: Amyloid β; Autophagy; GNE myopathy; Mitophagy; Muscle atrophy; Sialic acid.
© 2022. The Author(s).
Conflict of interest statement
The author declare no competing interests.
Figures
References
-
- Pogoryelova O, Cammish P, Mansbach H, Argov Z, Nishino I, Skrinar A, Chan Y, Nafissi S, Shamshiri H, Kakkis E, Lochmüller H. Phenotypic stratification and genotype-phenotype correlation in a heterogeneous, international cohort of GNE myopathy patients: first report from the GNE myopathy Disease Monitoring Program, registry portion. Neuromuscul Disord : NMD. 2018;28(2):158–168. doi: 10.1016/j.nmd.2017.11.001. - DOI - PMC - PubMed
-
- Eisenberg I, Avidan N, Potikha T, Hochner H, Chen M, Olender T, Barash M, Shemesh M, Sadeh M, Grabov-Nardini G, Shmilevich I, Friedmann A, Karpati G, Bradley WG, Baumbach L, Lancet D, Asher EB, Beckmann JS, Argov Z, Mitrani-Rosenbaum S. The UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase gene is mutated in recessive hereditary inclusion body myopathy. Nat Genet. 2001;29(1):83–87. doi: 10.1038/ng718. - DOI - PubMed
-
- Hinderlich S, Stäsche R, Zeitler R, Reutter W. A bifunctional enzyme catalyzes the first two steps in N-acetylneuraminic acid biosynthesis of rat liver. Purification and characterization of UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase. J Biol Chem. 1997;272(39):24313–24318. doi: 10.1074/jbc.272.39.24313. - DOI - PubMed
-
- Krause S, Hinderlich S, Amsili S, Horstkorte R, Wiendl H, Argov Z, Mitrani-Rosenbaum S, Lochmüller H. Localization of UDP-GlcNAc 2-epimerase/ManAc kinase (GNE) in the Golgi complex and the nucleus of mammalian cells. Exp Cell Res. 2005;304(2):365–379. doi: 10.1016/j.yexcr.2004.11.010. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
