

Clinical and genomic signatures of SARS-CoV-2 Delta breakthrough infections in New York
- PMID: 35906172
- PMCID: PMC9323230
- DOI: 10.1016/j.ebiom.2022.104141
Clinical and genomic signatures of SARS-CoV-2 Delta breakthrough infections in New York
Abstract
Background: In 2021, Delta became the predominant SARS-CoV-2 variant worldwide. While vaccines have effectively prevented COVID-19 hospitalization and death, vaccine breakthrough infections increasingly occurred. The precise role of clinical and genomic determinants in Delta infections is not known, and whether they contributed to increased rates of breakthrough infections compared to unvaccinated controls.
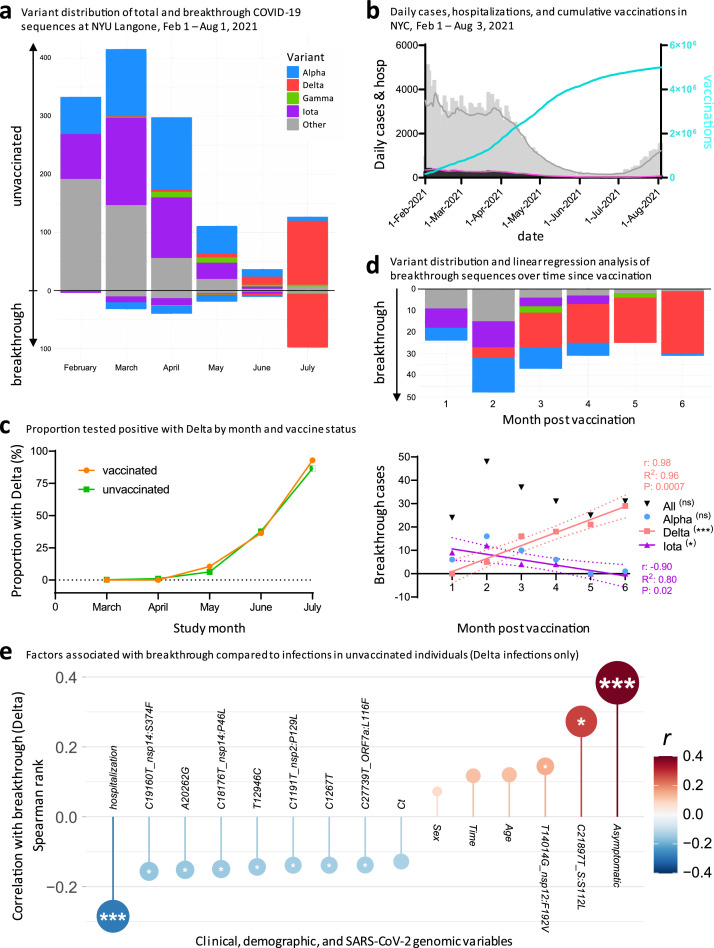
Methods: We studied SARS-CoV-2 variant distribution, dynamics, and adaptive selection over time in relation to vaccine status, phylogenetic relatedness of viruses, full genome mutation profiles, and associated clinical and demographic parameters.
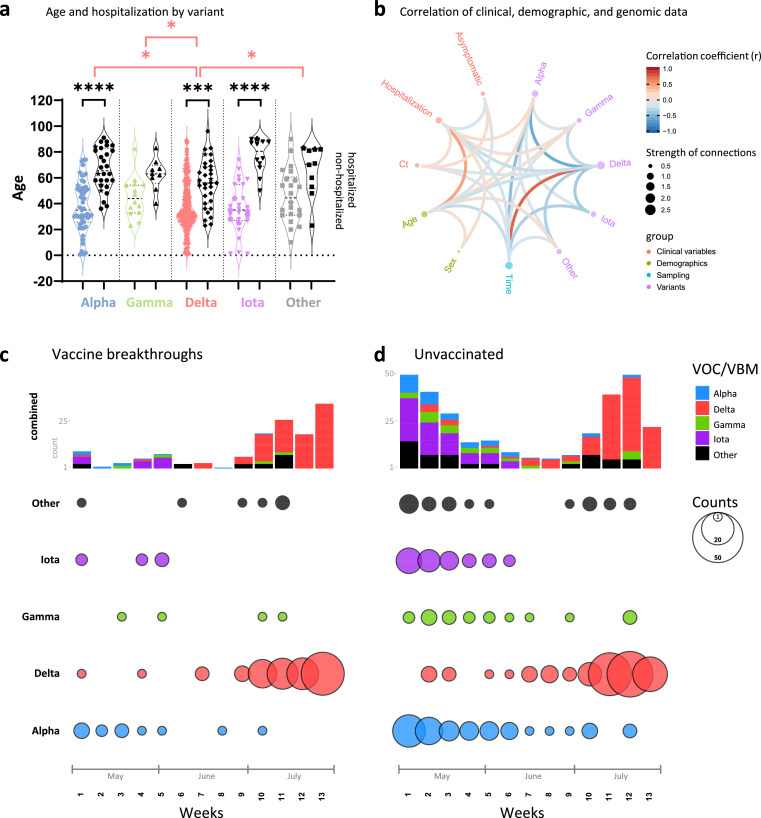
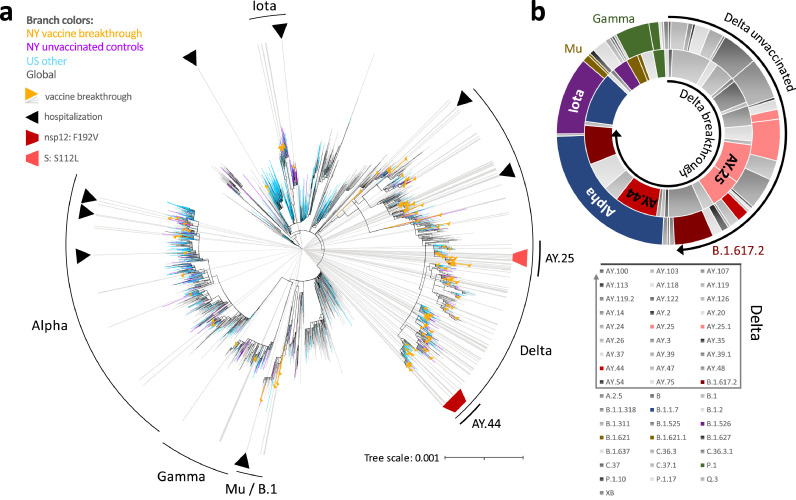
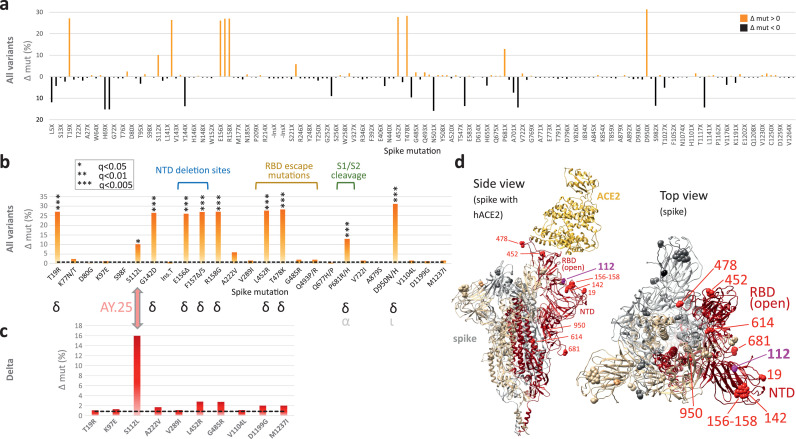
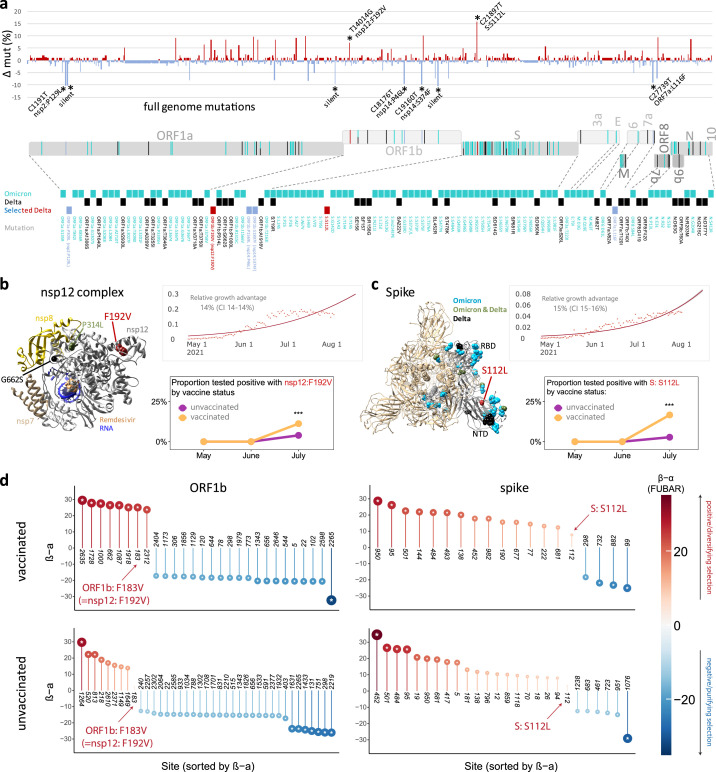
Findings: We show a steep and near-complete replacement of circulating variants with Delta between May and August 2021 in metropolitan New York. We observed an increase of the Delta sublineage AY.25 (14% in vaccinated, 7% in unvaccinated), its spike mutation S112L, and AY.44 (8% in vaccinated, 2% in unvaccinated) with its nsp12 mutation F192V in breakthroughs. Delta infections were associated with younger age and lower hospitalization rates than Alpha. Delta breakthrough infections increased significantly with time since vaccination, and, after adjusting for confounders, they rose at similar rates as in unvaccinated individuals.
Interpretation: We observed a modest adaptation of Delta genomes in breakthrough infections in New York, suggesting an improved genomic framework to support Delta's epidemic growth in times of waning vaccine protection despite limited impact on vaccine escape.
Funding: The study was supported by NYU institutional funds. The NYULH Genome Technology Center is partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center.
Keywords: Genomic signatures of vaccine breakthrough; Network analysis of clinical variables; SARS-CoV-2 variant of concern (VOC) delta; Selective adaptation; Spike S112L and nsp12 F192V; Time since vaccination.
Copyright © 2022 The Author(s). Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of interests The authors have no conflict of interest to declare.
Figures
Update of
-
Clinical and genomic signatures of rising SARS-CoV-2 Delta breakthrough infections in New York.medRxiv [Preprint]. 2021 Dec 17:2021.12.07.21267431. doi: 10.1101/2021.12.07.21267431. medRxiv. 2021. Update in: EBioMedicine. 2022 Aug;82:104141. doi: 10.1016/j.ebiom.2022.104141. PMID: 34909779 Free PMC article. Updated. Preprint.
References
-
- World Health Organization (WHO) 2021. WHO Coronavirus (COVID-19) Dashboard.https://covid19.who.int/ Accessed 3 October 2021.
-
- Centers for Disease Control and Prevention (CDC) 2021. SARS-CoV-2 Variant Classifications and Definitions.https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html Accessed 3 October 2021.
-
- GISAID . 2021. GISAID Database.https://www.gisaid.org/ Accessed 25 September 2021.
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
