

Cross-validation of distance measurements in proteins by PELDOR/DEER and single-molecule FRET
- PMID: 35906222
- PMCID: PMC9338047
- DOI: 10.1038/s41467-022-31945-6
Cross-validation of distance measurements in proteins by PELDOR/DEER and single-molecule FRET
Abstract
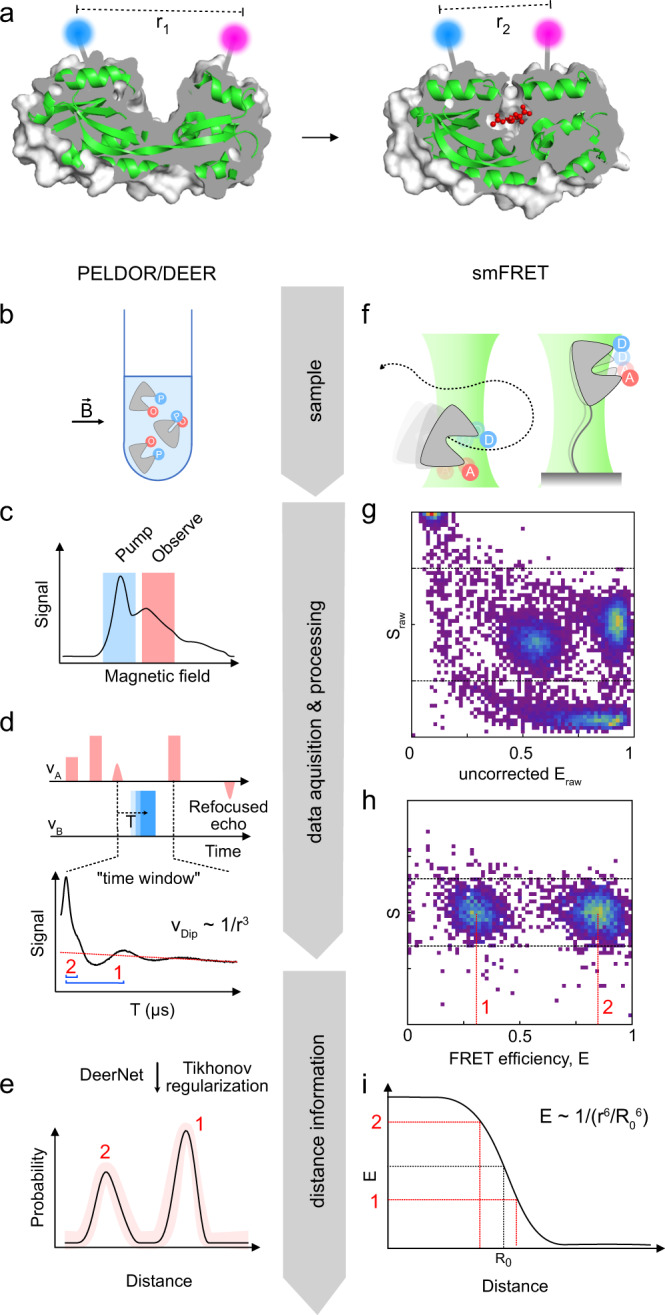
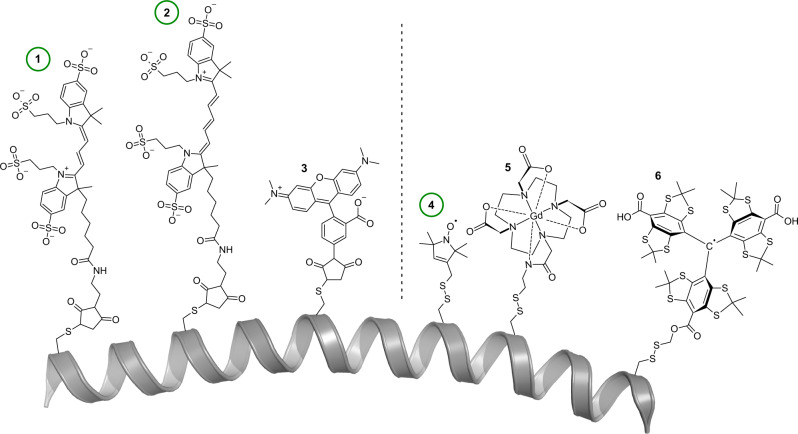
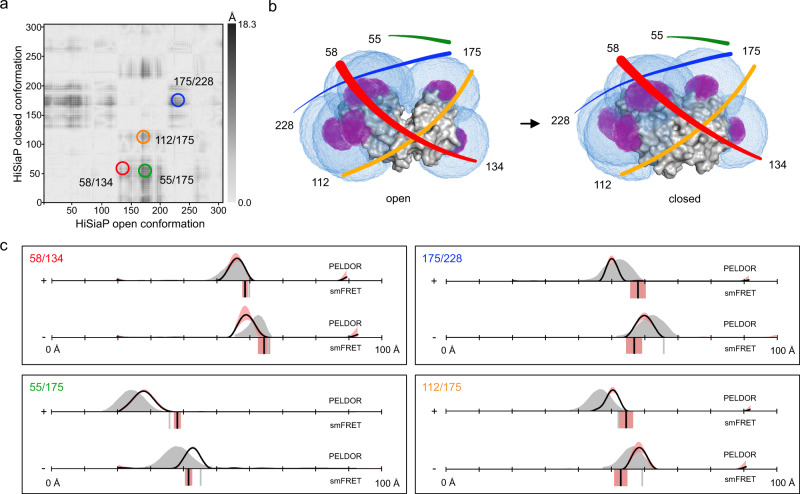
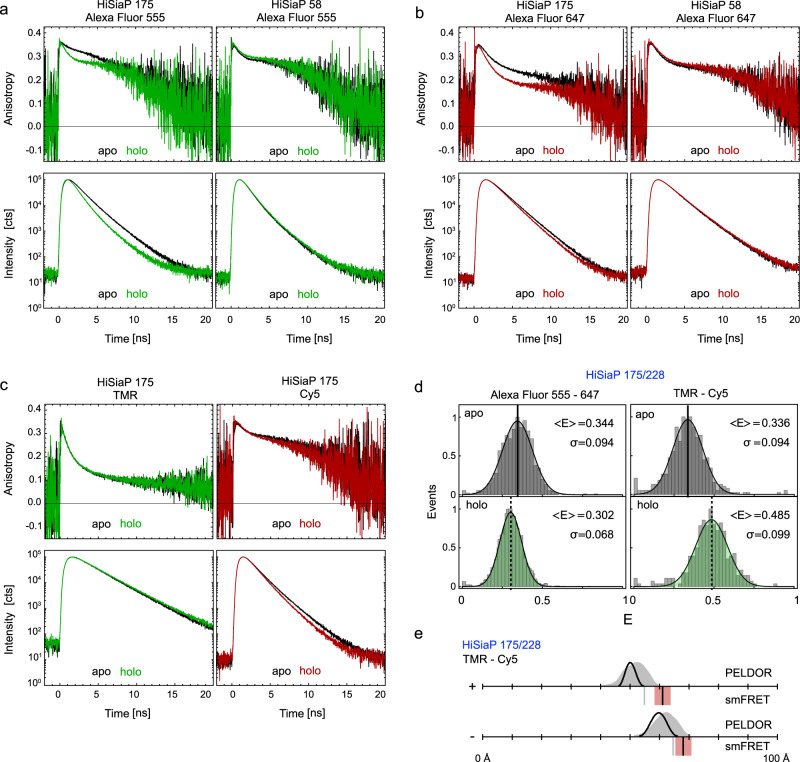
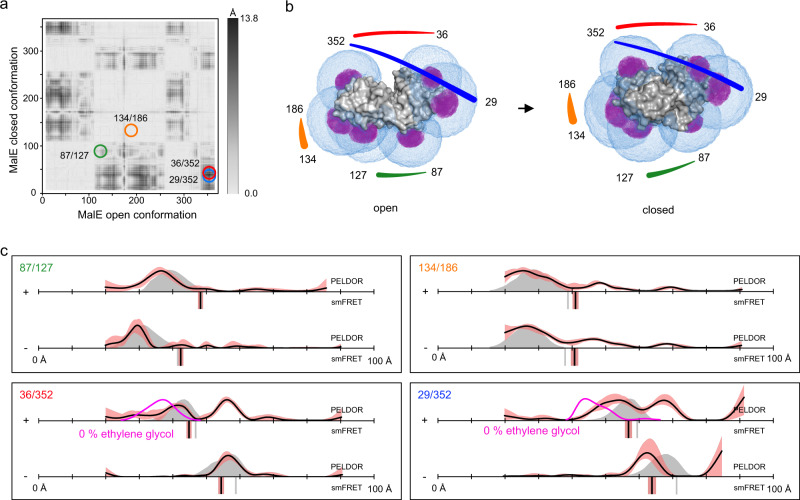
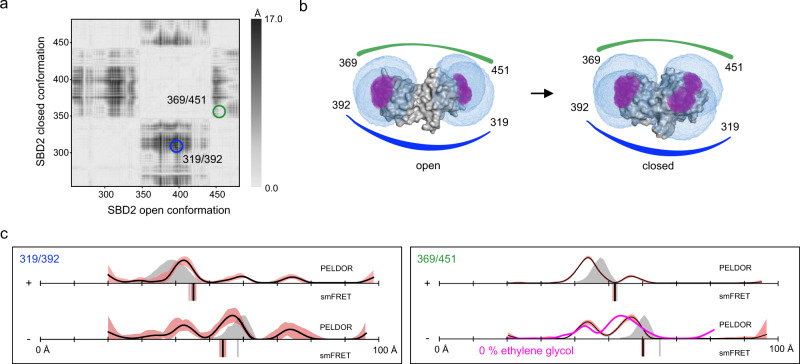
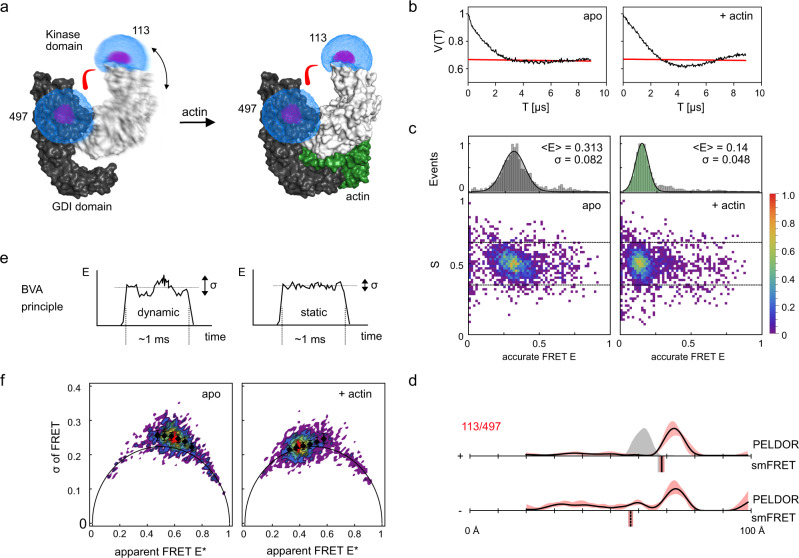
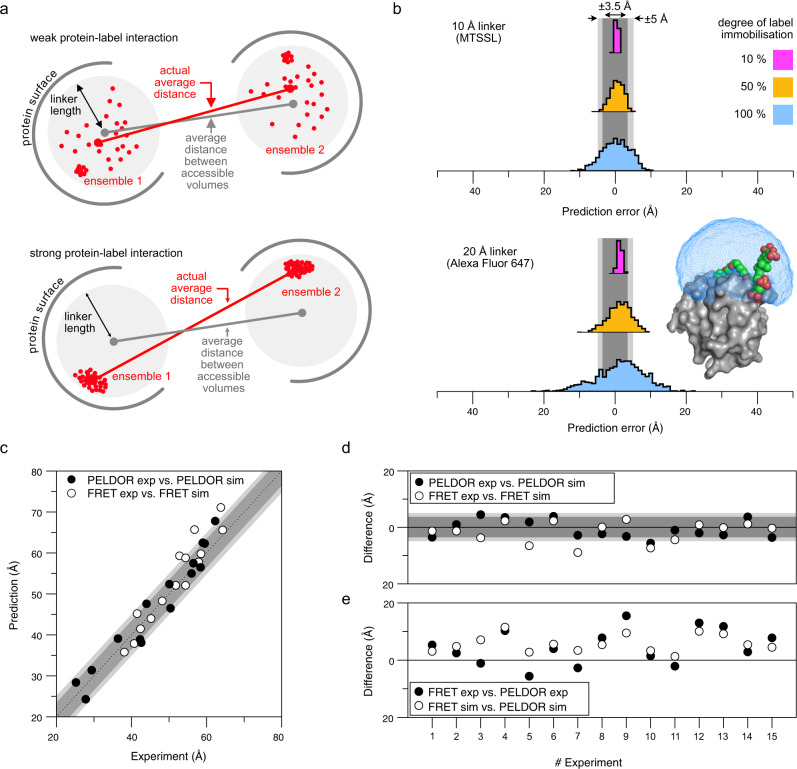
Pulsed electron-electron double resonance spectroscopy (PELDOR/DEER) and single-molecule Förster resonance energy transfer spectroscopy (smFRET) are frequently used to determine conformational changes, structural heterogeneity, and inter probe distances in biological macromolecules. They provide qualitative information that facilitates mechanistic understanding of biochemical processes and quantitative data for structural modelling. To provide a comprehensive comparison of the accuracy of PELDOR/DEER and smFRET, we use a library of double cysteine variants of four proteins that undergo large-scale conformational changes upon ligand binding. With either method, we use established standard experimental protocols and data analysis routines to determine inter-probe distances in the presence and absence of ligands. The results are compared to distance predictions from structural models. Despite an overall satisfying and similar distance accuracy, some inconsistencies are identified, which we attribute to the use of cryoprotectants for PELDOR/DEER and label-protein interactions for smFRET. This large-scale cross-validation of PELDOR/DEER and smFRET highlights the strengths, weaknesses, and synergies of these two important and complementary tools in integrative structural biology.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
