

X-CAP improves pathogenicity prediction of stopgain variants
- PMID: 35906703
- PMCID: PMC9338606
- DOI: 10.1186/s13073-022-01078-y
X-CAP improves pathogenicity prediction of stopgain variants
Abstract
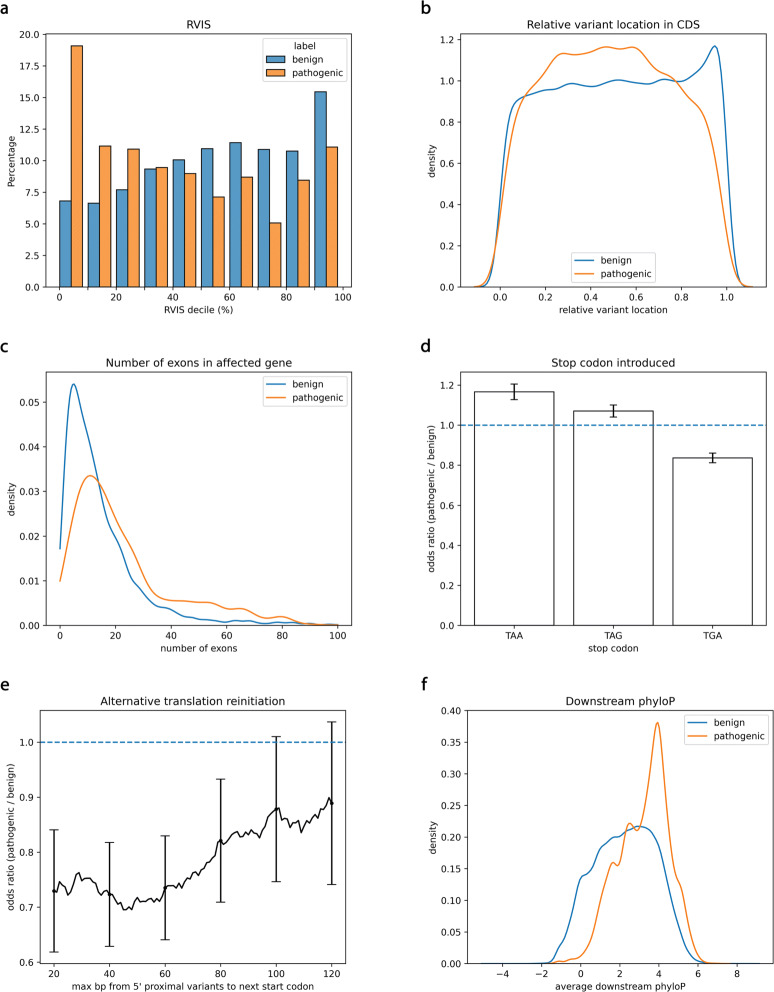
Stopgain substitutions are the third-largest class of monogenic human disease mutations and often examined first in patient exomes. Existing computational stopgain pathogenicity predictors, however, exhibit poor performance at the high sensitivity required for clinical use. Here, we introduce a new classifier, termed X-CAP, which uses a novel training methodology and unique feature set to improve the AUROC by 18% and decrease the false-positive rate 4-fold on large variant databases. In patient exomes, X-CAP prioritizes causal stopgains better than existing methods do, further illustrating its clinical utility. X-CAP is available at https://github.com/bejerano-lab/X-CAP .
Keywords: Machine learning; Nonsense; Pathogenicity prediction; Stopgain.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
-
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, et al. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet. 2020;139(10):1197–207. doi: 10.1007/s00439-020-02199-3. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
