

ADGRL1 haploinsufficiency causes a variable spectrum of neurodevelopmental disorders in humans and alters synaptic activity and behavior in a mouse model
- PMID: 35907405
- PMCID: PMC9388395
- DOI: 10.1016/j.ajhg.2022.06.011
ADGRL1 haploinsufficiency causes a variable spectrum of neurodevelopmental disorders in humans and alters synaptic activity and behavior in a mouse model
Abstract
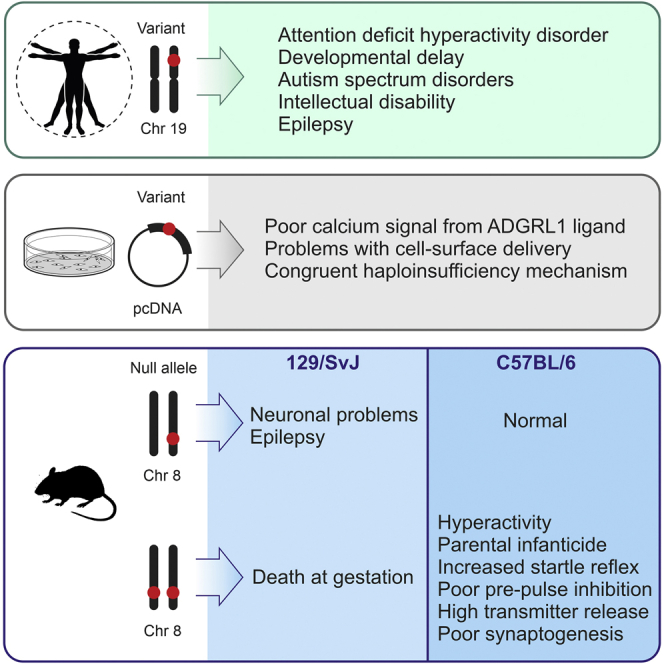
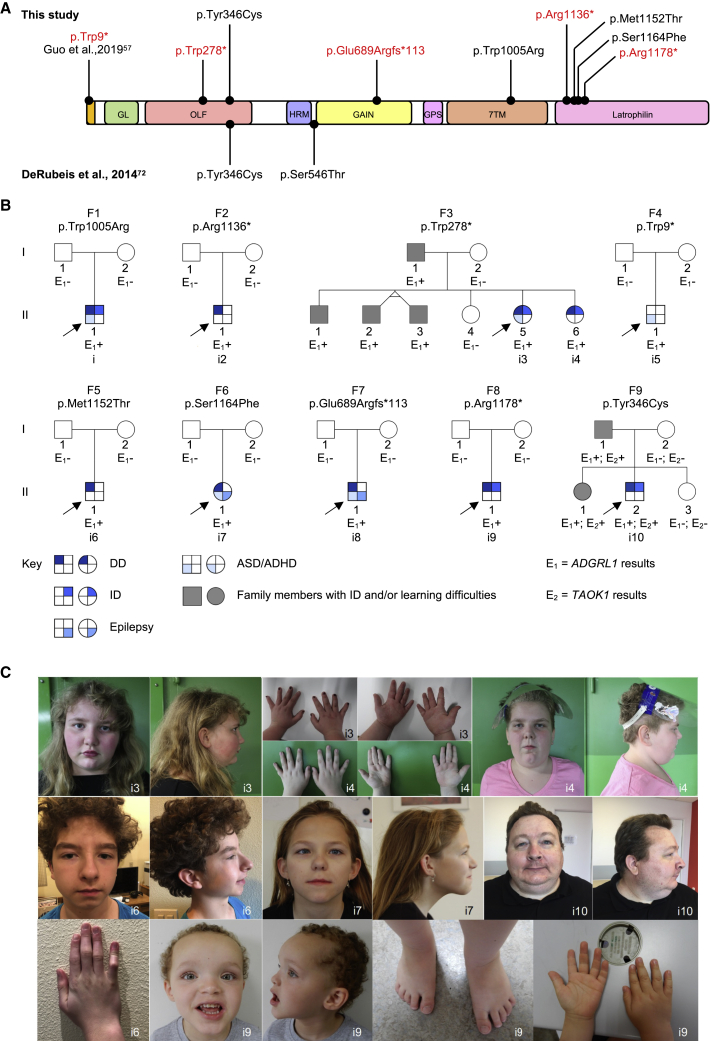
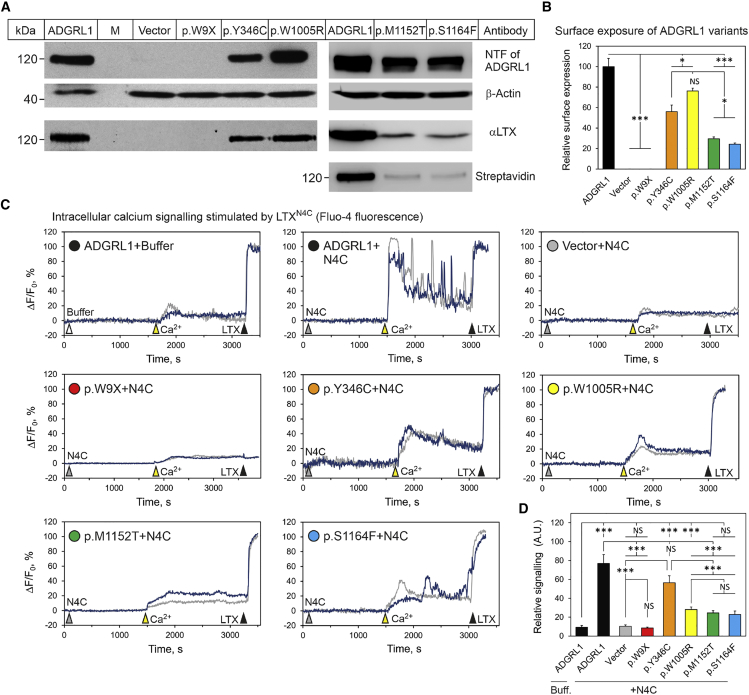
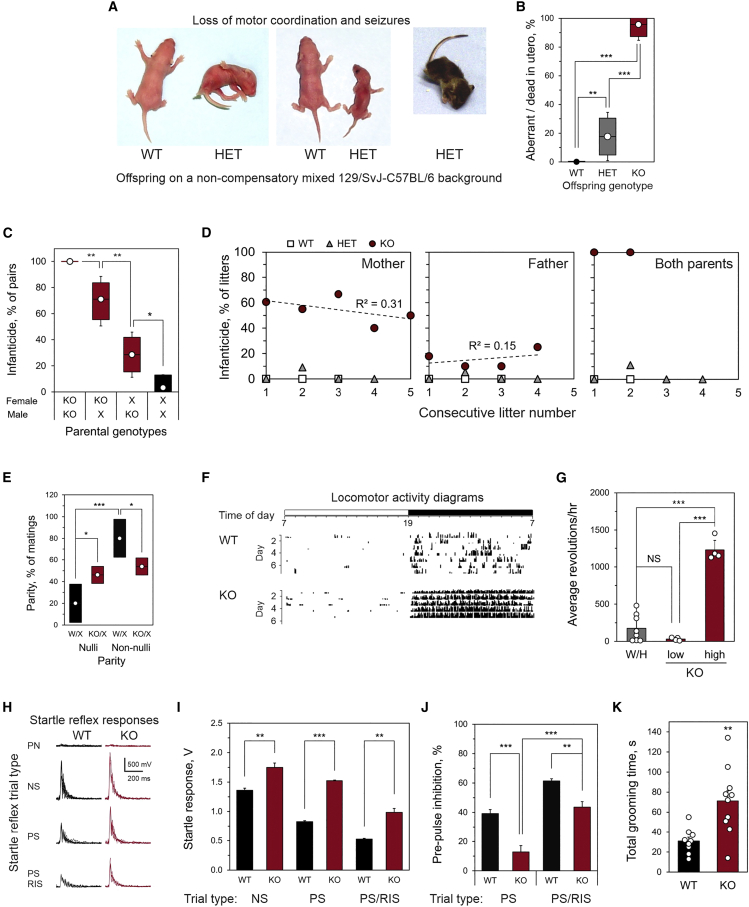
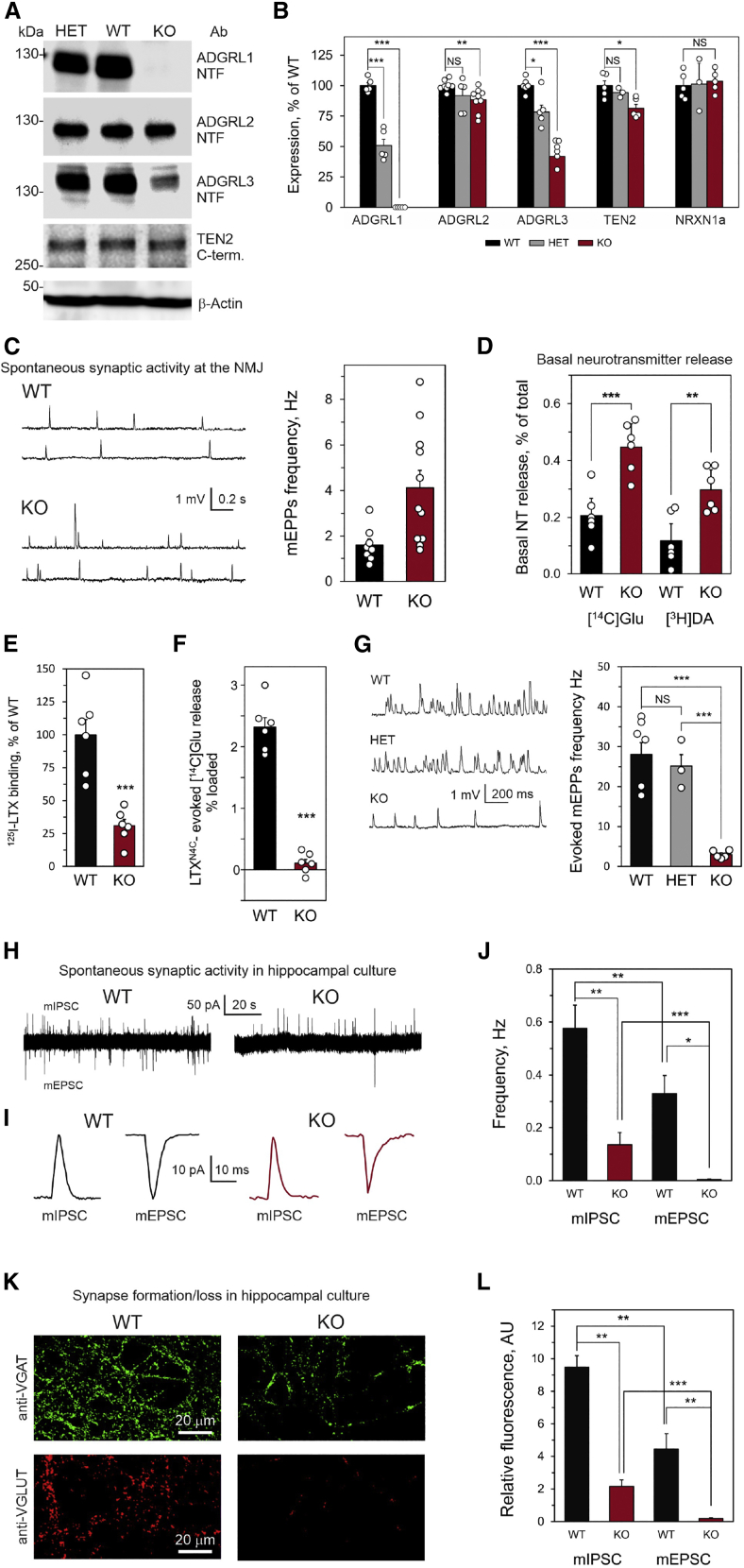
ADGRL1 (latrophilin 1), a well-characterized adhesion G protein-coupled receptor, has been implicated in synaptic development, maturation, and activity. However, the role of ADGRL1 in human disease has been elusive. Here, we describe ten individuals with variable neurodevelopmental features including developmental delay, intellectual disability, attention deficit hyperactivity and autism spectrum disorders, and epilepsy, all heterozygous for variants in ADGRL1. In vitro, human ADGRL1 variants expressed in neuroblastoma cells showed faulty ligand-induced regulation of intracellular Ca2+ influx, consistent with haploinsufficiency. In vivo, Adgrl1 was knocked out in mice and studied on two genetic backgrounds. On a non-permissive background, mice carrying a heterozygous Adgrl1 null allele exhibited neurological and developmental abnormalities, while homozygous mice were non-viable. On a permissive background, knockout animals were also born at sub-Mendelian ratios, but many Adgrl1 null mice survived gestation and reached adulthood. Adgrl1-/- mice demonstrated stereotypic behaviors, sexual dysfunction, bimodal extremes of locomotion, augmented startle reflex, and attenuated pre-pulse inhibition, which responded to risperidone. Ex vivo synaptic preparations displayed increased spontaneous exocytosis of dopamine, acetylcholine, and glutamate, but Adgrl1-/- neurons formed synapses in vitro poorly. Overall, our findings demonstrate that ADGRL1 haploinsufficiency leads to consistent developmental, neurological, and behavioral abnormalities in mice and humans.
Keywords: ADGRL1; ADHD; ASD; Adgrl1 knockout mice; attention deficit hyperactivity disorder; autism spectrum disorder; developmental delay; epilepsy; intellectual disability; malfunctional behavior in mice; neuropsychiatric disorders; variable expressivity.
Copyright © 2022 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories. A.C. is an employee of GeneDx, Inc.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
