

Neuroinflammation: Extinguishing a blaze of T cells
- PMID: 35909230
- PMCID: PMC9489683
- DOI: 10.1111/imr.13122
Neuroinflammation: Extinguishing a blaze of T cells
Abstract
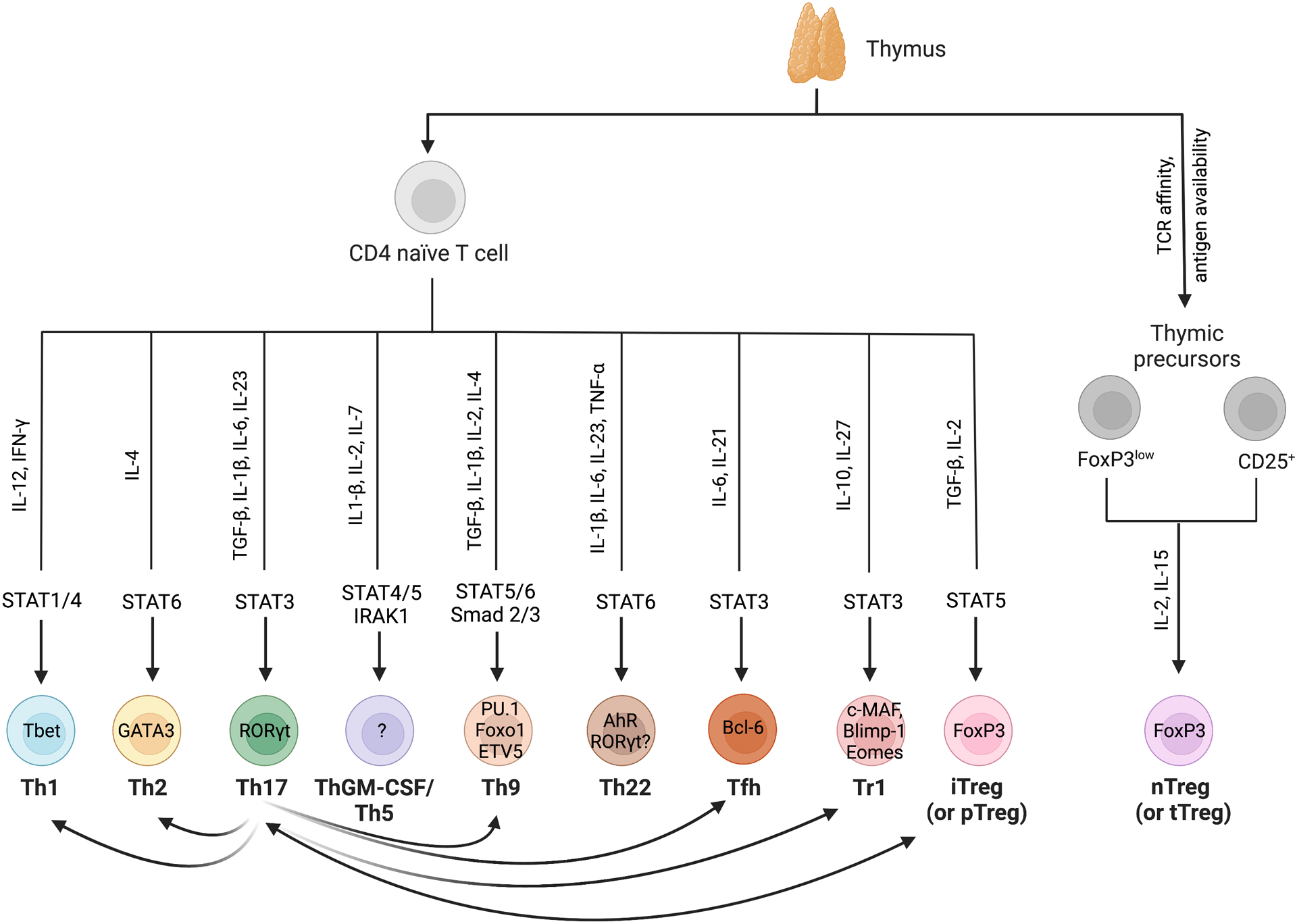
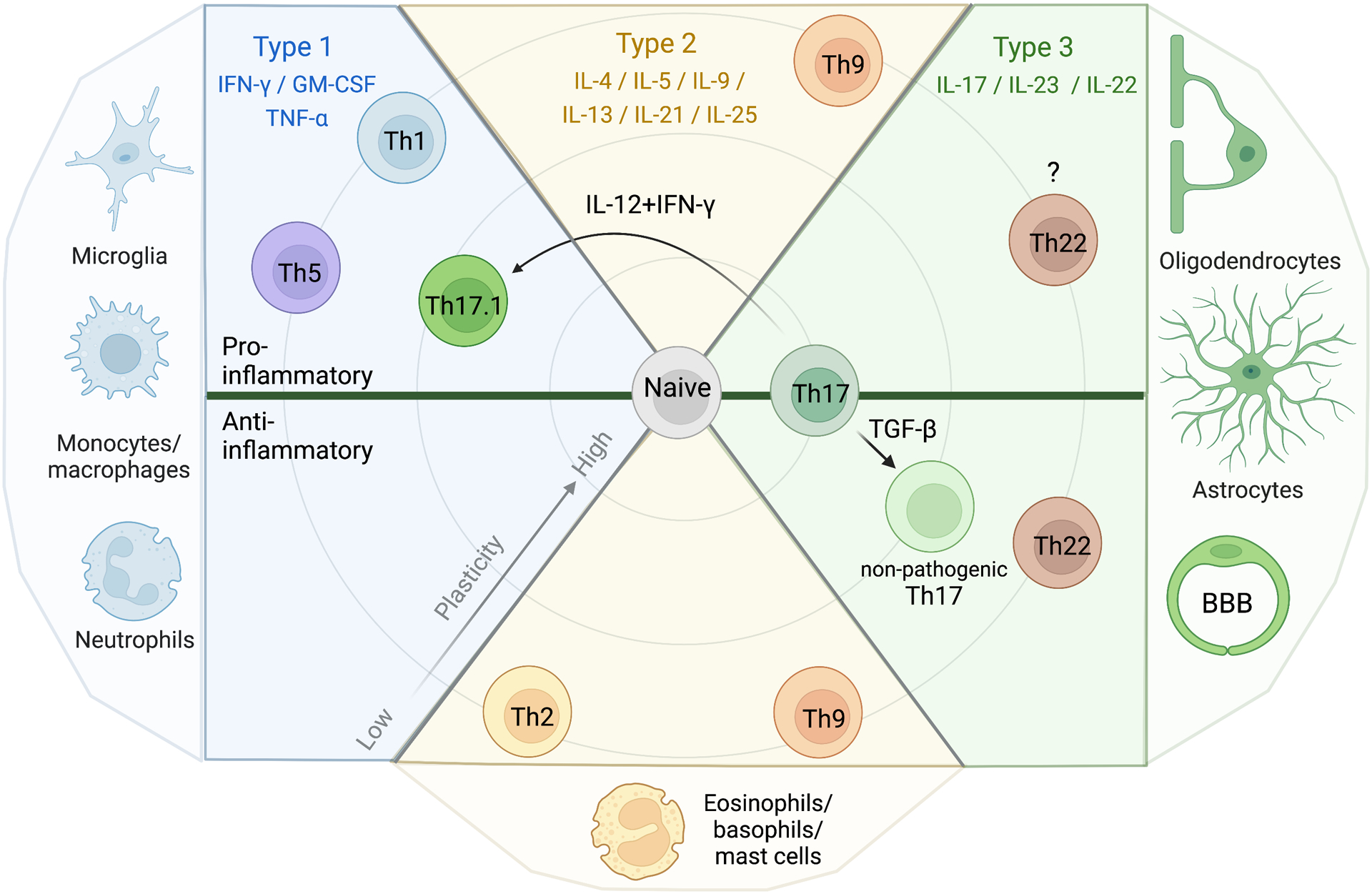
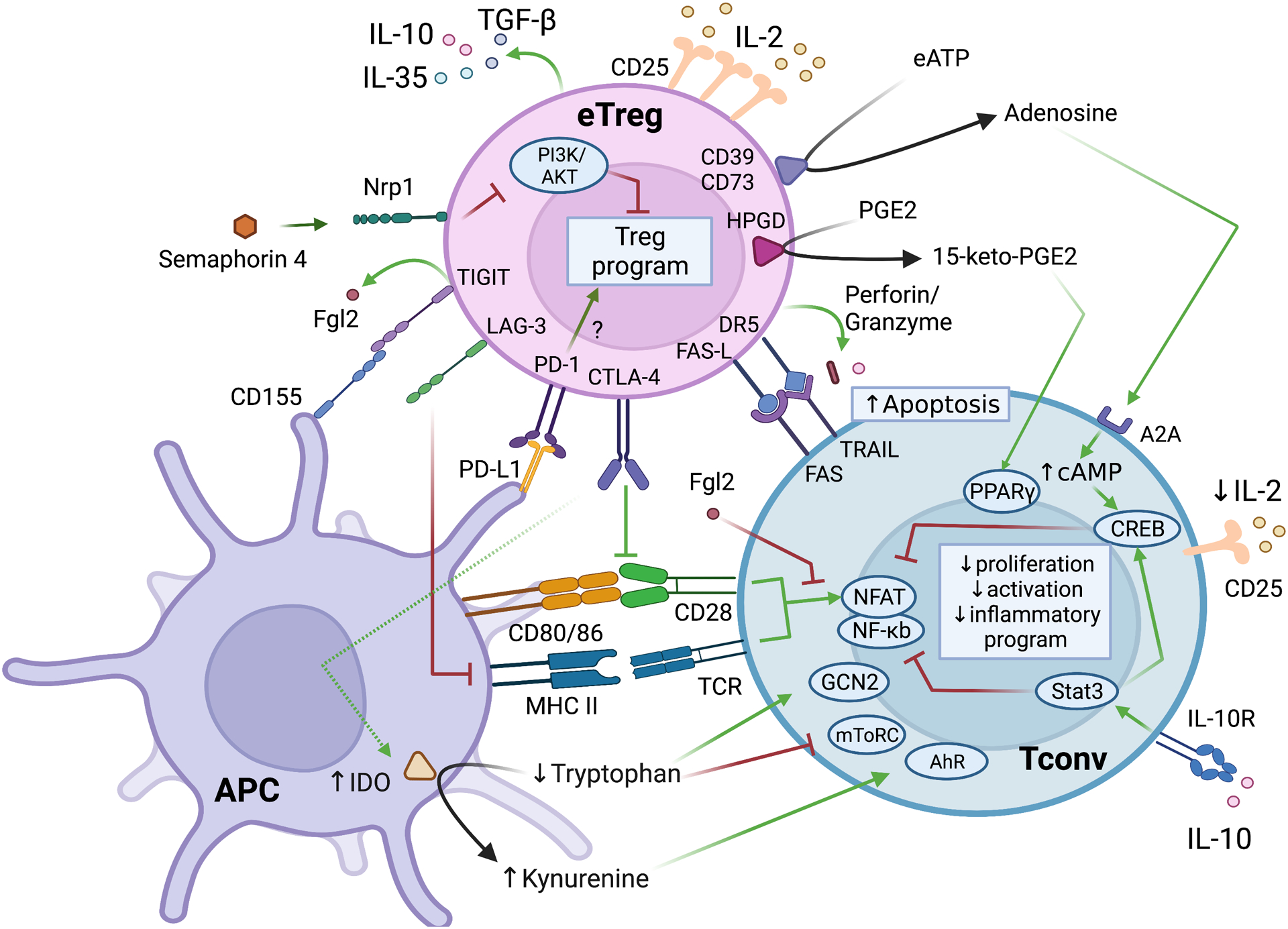
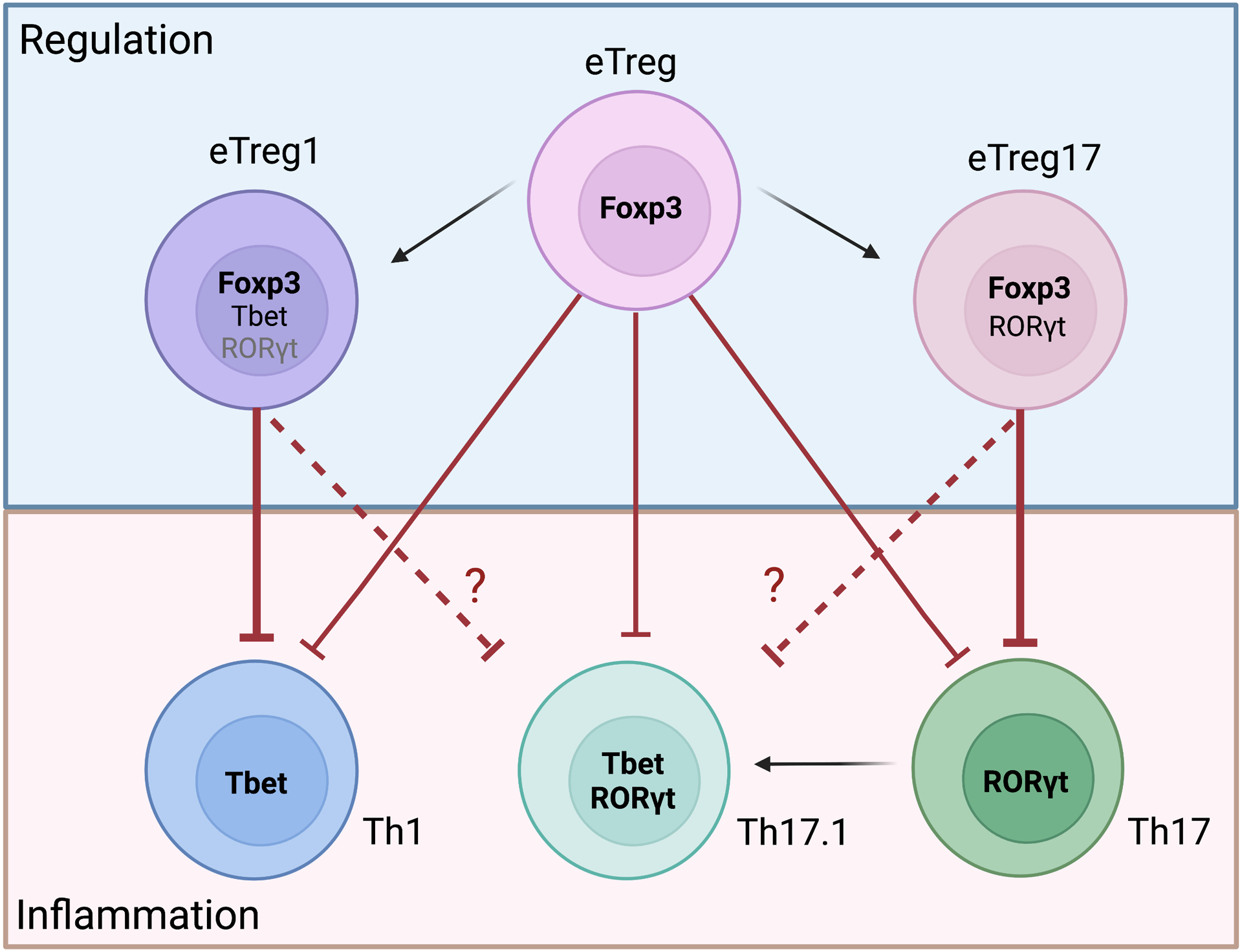
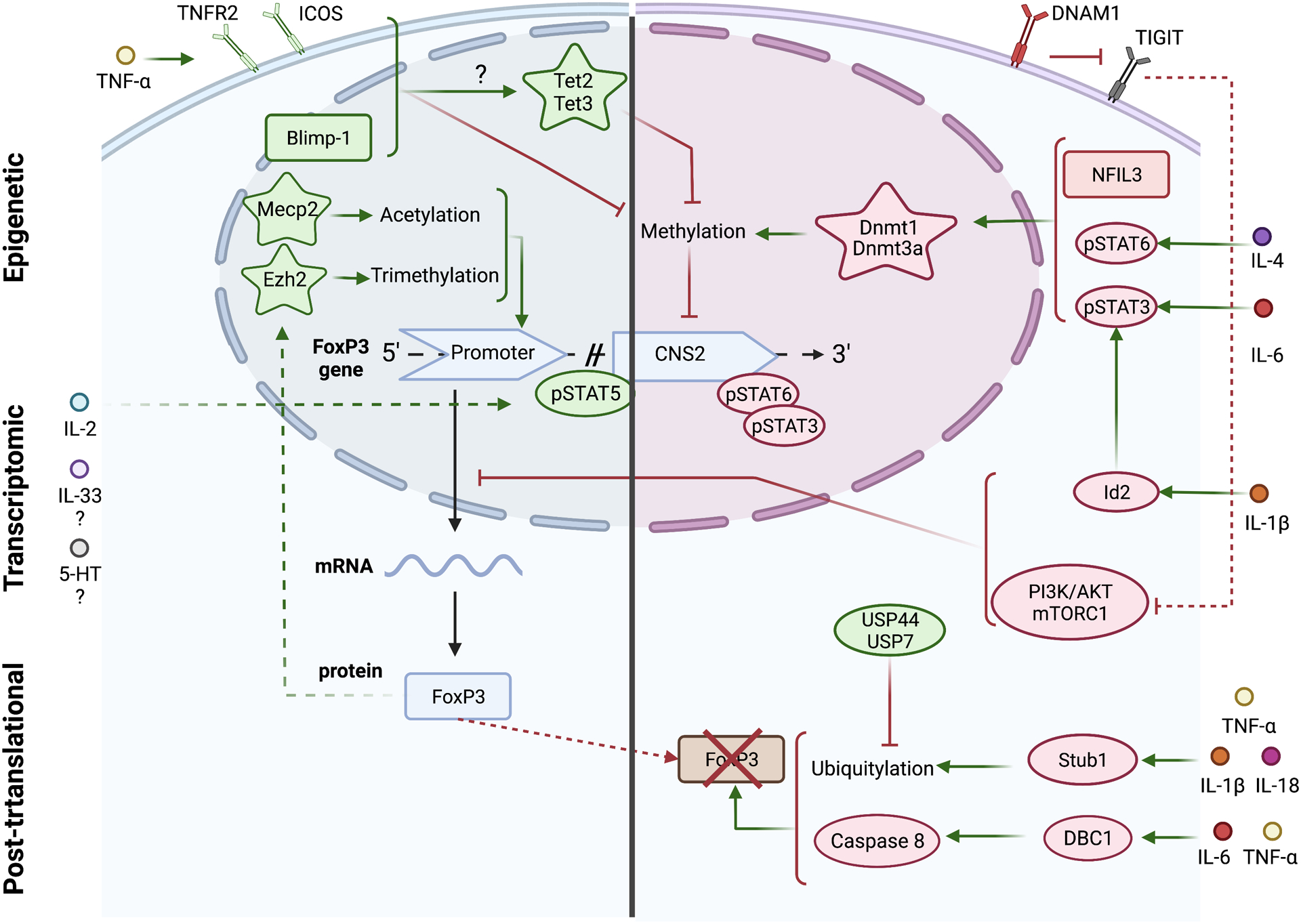
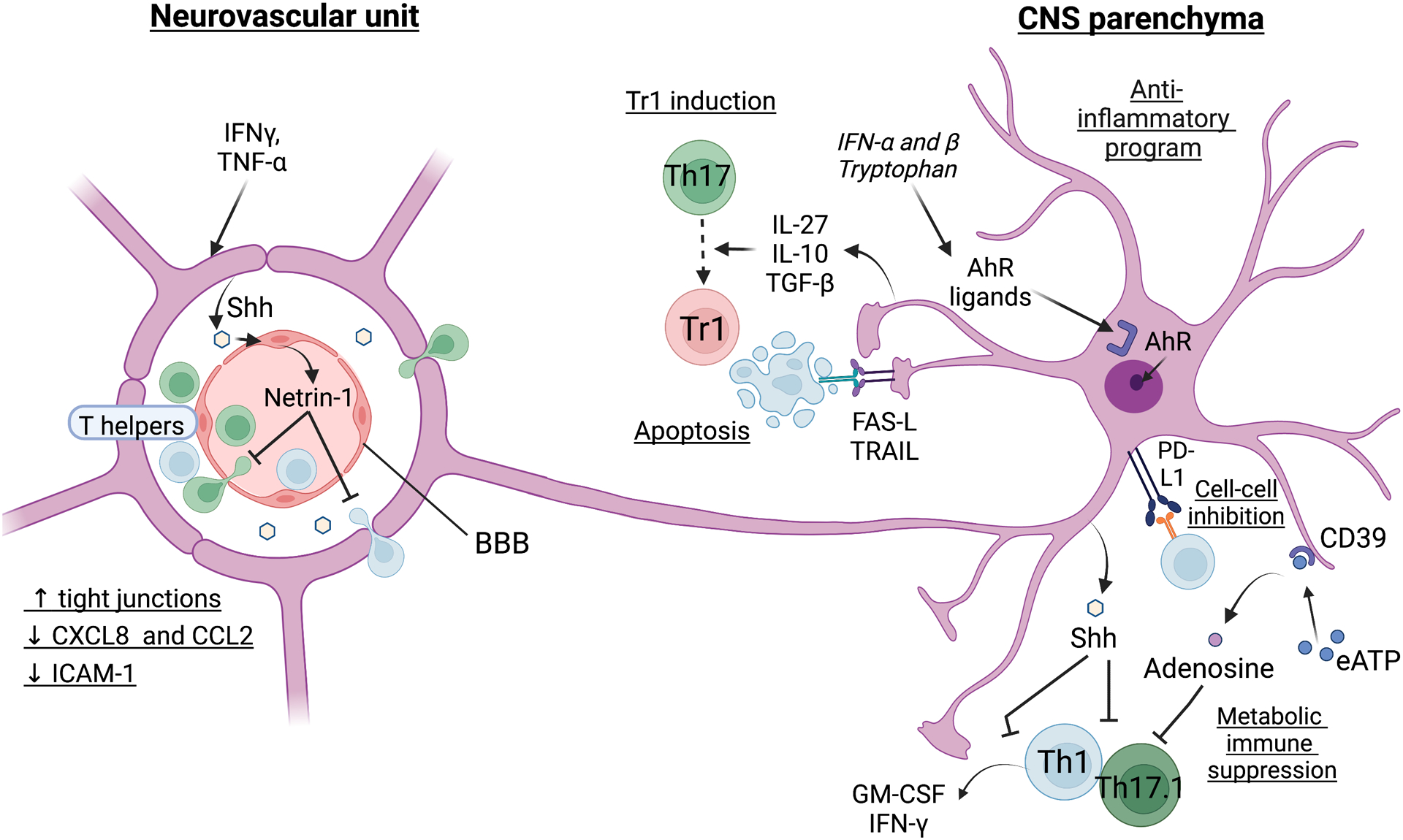
Inflammation is a biological process that dynamically alters the surrounding microenvironment, including participating immune cells. As a well-protected organ surrounded by specialized barriers and with immune privilege properties, the central nervous system (CNS) tightly regulates immune responses. Yet in neuroinflammatory conditions, pathogenic immunity can disrupt CNS structure and function. T cells in particular play a key role in promoting and restricting neuroinflammatory responses, while the inflamed CNS microenvironment can influence and reshape T cell function and identity. Still, the contraction of aberrant T cell responses within the CNS is not well understood. Using autoimmunity as a model, here we address the contribution of CD4 T helper (Th) cell subsets in promoting neuropathology and disease. To address the mechanisms antagonizing neuroinflammation, we focus on the control of the immune response by regulatory T cells (Tregs) and describe the counteracting processes that preserve their identity under inflammatory challenges. Finally, given the influence of the local microenvironment on immune regulation, we address how CNS-intrinsic signals reshape T cell function to mitigate abnormal immune T cell responses.
Keywords: T cells; Th cells; Tregs; autoimmunity; central nervous system; inflammation.
© 2022 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors have no conflict of interest to disclose.
Figures
Similar articles
-
Regulatory T cells selectively preserve immune privilege of self-antigens during viral central nervous system infection.J Immunol. 2012 Apr 15;188(8):3678-85. doi: 10.4049/jimmunol.1102422. Epub 2012 Mar 9. J Immunol. 2012. PMID: 22407917
-
Metabolic regulation and function of T helper cells in neuroinflammation.Semin Immunopathol. 2022 Sep;44(5):581-598. doi: 10.1007/s00281-022-00959-z. Epub 2022 Sep 6. Semin Immunopathol. 2022. PMID: 36068310 Review.
-
Human CD4+ T cell subsets differ in their abilities to cross endothelial and epithelial brain barriers in vitro.Fluids Barriers CNS. 2020 Feb 3;17(1):3. doi: 10.1186/s12987-019-0165-2. Fluids Barriers CNS. 2020. PMID: 32008573 Free PMC article.
-
The CXCL13/CXCR5-chemokine axis in neuroinflammation: evidence of CXCR5+CD4 T cell recruitment to CSF.Fluids Barriers CNS. 2021 Aug 26;18(1):40. doi: 10.1186/s12987-021-00272-1. Fluids Barriers CNS. 2021. PMID: 34446066 Free PMC article.
-
Regulation of lymphocyte trafficking in central nervous system autoimmunity.Curr Opin Immunol. 2018 Dec;55:38-43. doi: 10.1016/j.coi.2018.09.008. Epub 2018 Sep 27. Curr Opin Immunol. 2018. PMID: 30268837 Free PMC article. Review.
Cited by
-
An in silico approach to identify early damage biomarker candidates in metachromatic leukodystrophy.Mol Genet Metab Rep. 2023 May 15;35:100974. doi: 10.1016/j.ymgmr.2023.100974. eCollection 2023 Jun. Mol Genet Metab Rep. 2023. PMID: 37275681 Free PMC article.
-
Berberine promotes immunological outcomes and decreases neuroinflammation in the experimental model of multiple sclerosis through the expansion of Treg and Th2 cells.Immun Inflamm Dis. 2023 Jan;11(1):e766. doi: 10.1002/iid3.766. Immun Inflamm Dis. 2023. PMID: 36705421 Free PMC article.
-
An introduction to neuroimmunology.Immunol Rev. 2022 Oct;311(1):5-8. doi: 10.1111/imr.13133. Epub 2022 Aug 30. Immunol Rev. 2022. PMID: 36039857 Free PMC article. No abstract available.
-
Neuroinflammation, memory, and depression: new approaches to hippocampal neurogenesis.J Neuroinflammation. 2023 Nov 27;20(1):283. doi: 10.1186/s12974-023-02964-x. J Neuroinflammation. 2023. PMID: 38012702 Free PMC article. Review.
References
-
- Dendrou CA, Fugger L, Friese MA. Immunopathology of multiple sclerosis. Nature reviews Immunology. 2015;15(9):545–558. - PubMed
-
- Hayashi T, Morimoto C, Burks JS, Kerr C, Hauser SL. Dual-label immunocytochemistry of the active multiple sclerosis lesion: major histocompatibility complex and activation antigens. Annals of neurology. 1988;24(4):523–531. - PubMed
-
- Booss J, Esiri MM, Tourtellotte WW, Mason DY. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. Journal of the neurological sciences. 1983;62(1–3):219–232. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
