

Comparing Clinical and Genetic Characteristics of De Novo and Inherited COL1A1/COL1A2 Variants in a Large Chinese Cohort of Osteogenesis Imperfecta
- PMID: 35909573
- PMCID: PMC9329653
- DOI: 10.3389/fendo.2022.935905
Comparing Clinical and Genetic Characteristics of De Novo and Inherited COL1A1/COL1A2 Variants in a Large Chinese Cohort of Osteogenesis Imperfecta
Abstract
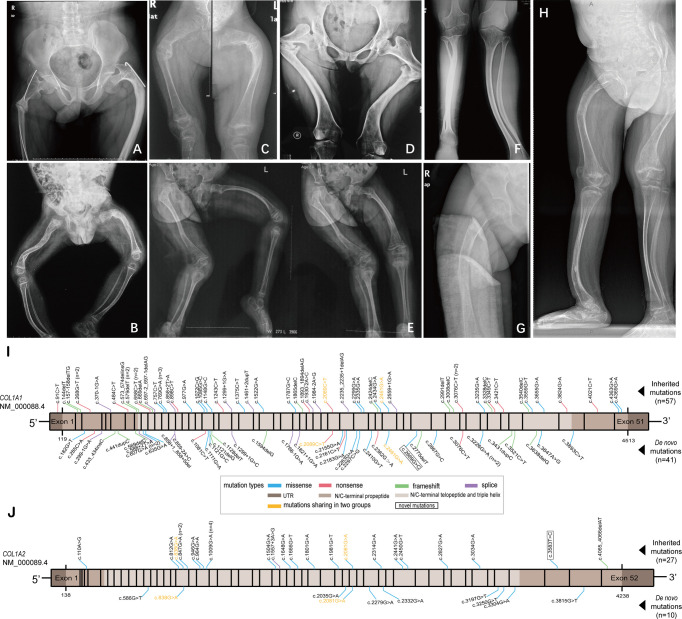
Purpose: Nearly 85%-90% of osteogenesis imperfecta (OI) cases are caused by autosome dominant mutations of COL1A1 and COL1A2 genes, of which de novo mutations cover a large proportion, whereas their characteristics remain to be elucidated. This study aims to compare the differences in clinical and genetic characteristics of de novo and inherited COL1A1/COL1A2 mutations of OI, assess the average paternal and maternal age at conception in de novo mutations, and research the rate of nonpenetrance in inherited mutations.
Materials and methods: A retrospective comparison between de novo and inherited mutations was performed among 135 OI probands with COL1A1/COL1A2 mutations. Mutational analyses of all probands and their family members were completed by Sanger sequencing. A new clinical scoring system was developed to assess the clinical severity of OI quantitatively.
Results: A total of 51 probands (37.78%) with de novo mutations and 84 probands (62.22%) with inherited mutations were grouped by the results of the parental gene verification. The proportion of clinical type III (P<0.001) and clinical scores (P<0.001) were significantly higher in de novo mutations. Missense mutations covered a slightly higher proportion of de novo COL1A1 mutations (46.34%) compared with inherited COL1A1 mutations (33.33%), however, lacking a significant difference (P=0.1923). The mean BMD Z/T-score at the lumbar spine in de novo mutations was -2.3 ± 1.5, lower than inherited mutations (-1.7 ± 1.8), but lacking statistical significance (P=0.0742). There was no significant difference between the two groups in OI-related phenotypes (like fracture frequency, blue sclera, and hearing loss) and biochemical indexes. In de novo mutations, the average paternal and maternal age at conception was 29.2 (P<0.05) and 26.8 (P<0.0001), respectively, which were significantly younger than the average gestational age of the population. Additionally, 98.04% of pedigrees (50/51) with de novo mutations were spontaneous conception. The rate of nonpenetrance of parents with pathogenic variants in the inherited mutation group was 25.64% (20/78).
Conclusions: Our data revealed that the proportion of clinical type III and clinical scores were significantly higher in de novo mutations than in inherited mutations, demonstrating that de novo mutations are more damaging because they have not undergone purifying selection.
Keywords: COL1A1; COL1A2; clinical scoring system; de novo; inherited; osteogenesis imperfecta.
Copyright © 2022 Mei, Zhang and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures


References
-
- Marini JC, Forlino A, Cabral WA, Barnes AM, San Antonio JD, Milgrom S, et al. Consortium for Osteogenesis Imperfecta Mutations in the Helical Domain of Type I Collagen: Regions Rich in Lethal Mutations Align With Collagen Binding Sites for Integrins and Proteoglycans. Hum Mutat (2007) 28(3):209–21. doi: 10.1002/humu.20429 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous