

lncRNA HITT Inhibits Lactate Production by Repressing PKM2 Oligomerization to Reduce Tumor Growth and Macrophage Polarization
- PMID: 35909936
- PMCID: PMC9285634
- DOI: 10.34133/2022/9854904
lncRNA HITT Inhibits Lactate Production by Repressing PKM2 Oligomerization to Reduce Tumor Growth and Macrophage Polarization
Abstract
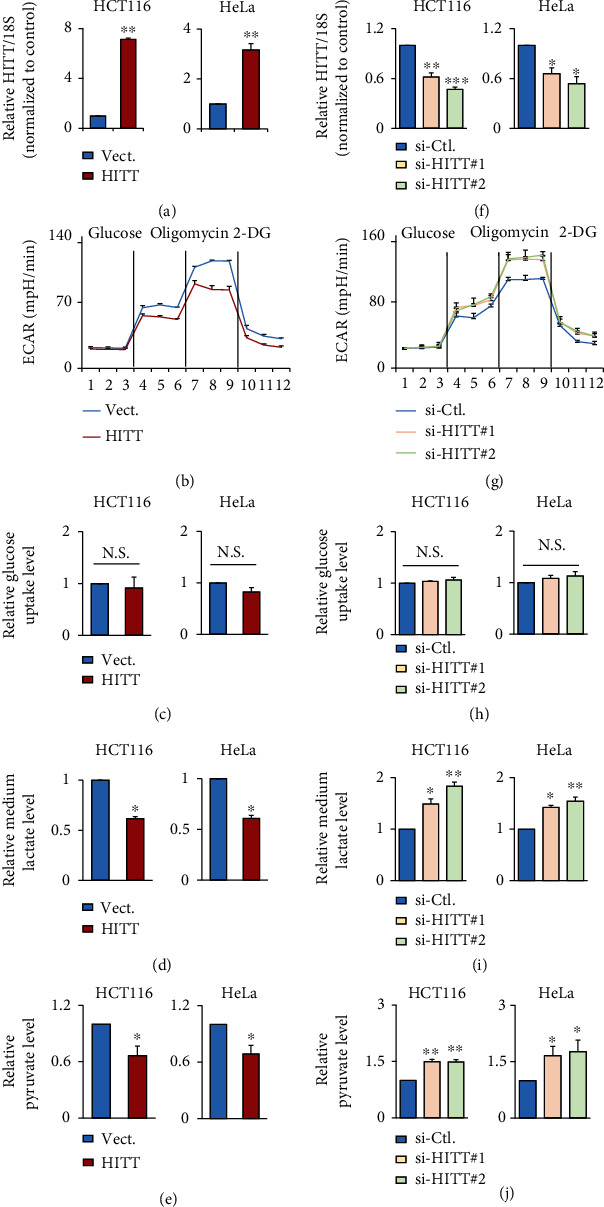
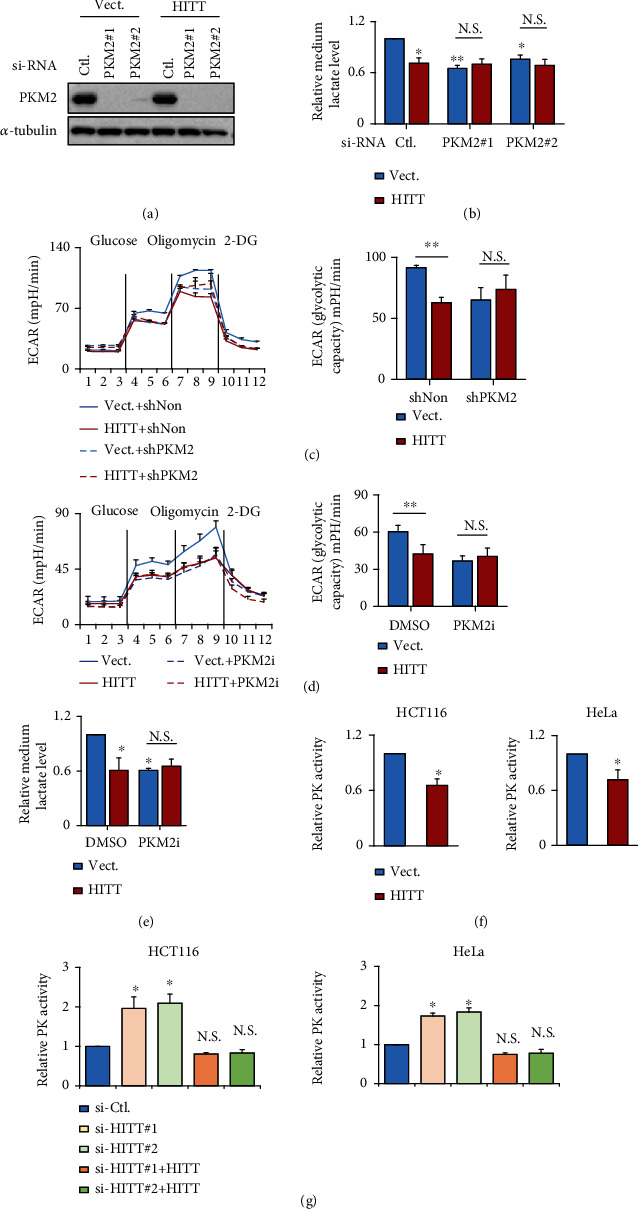
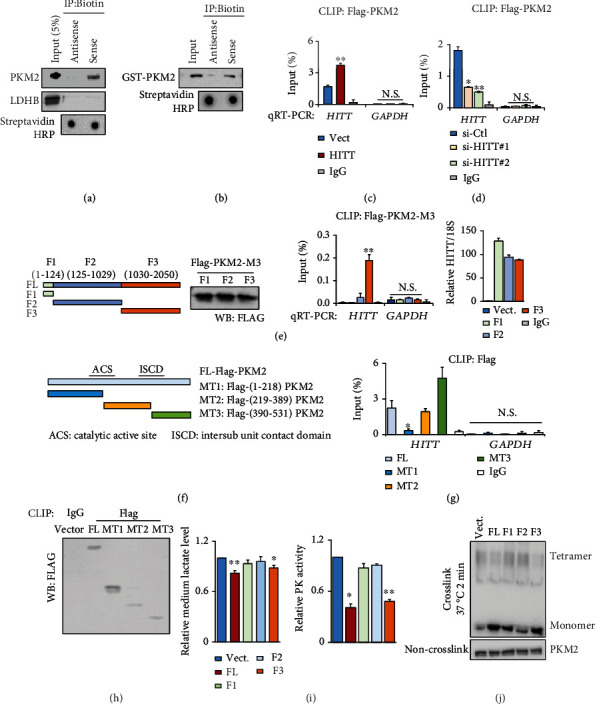
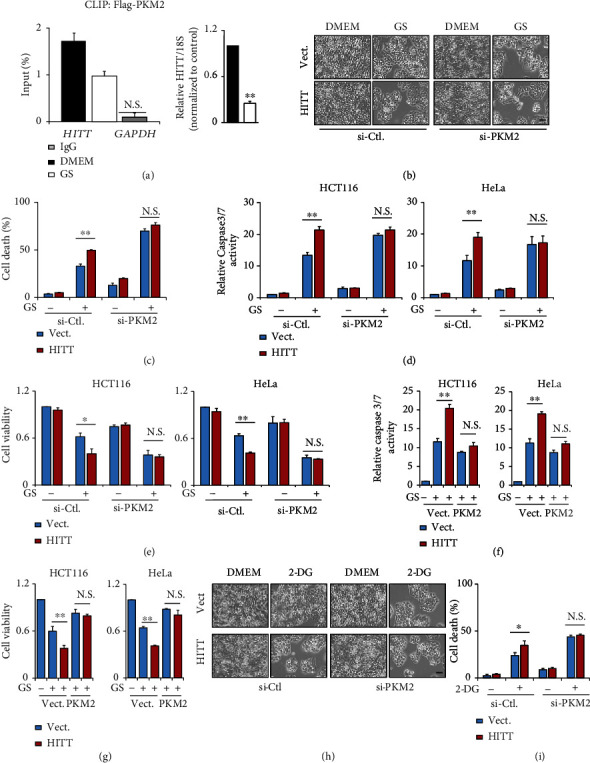
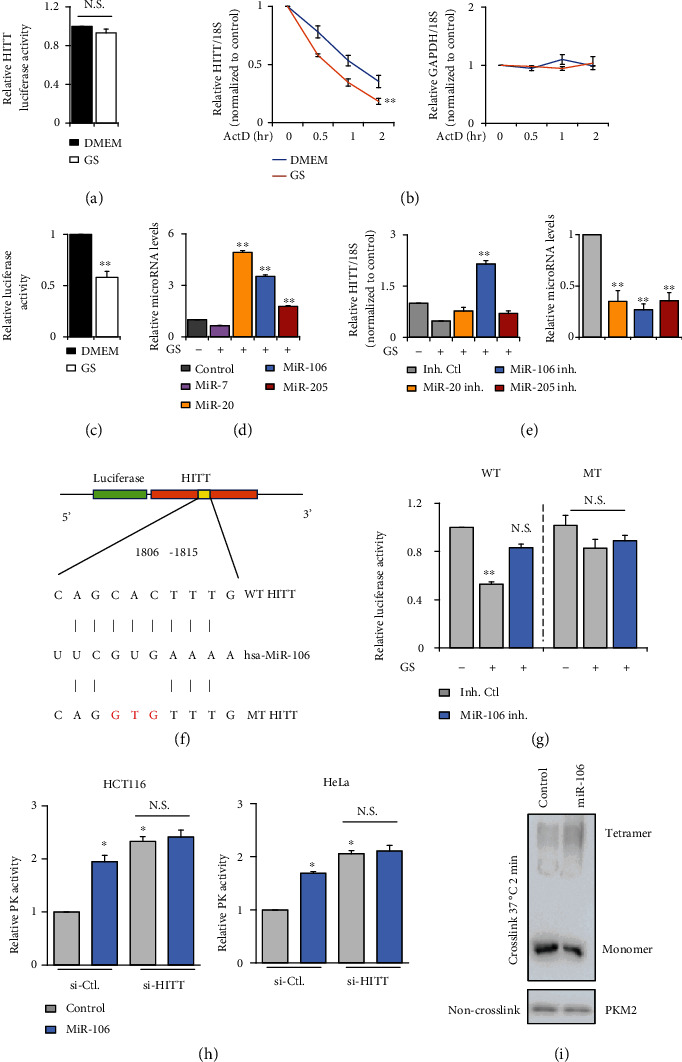
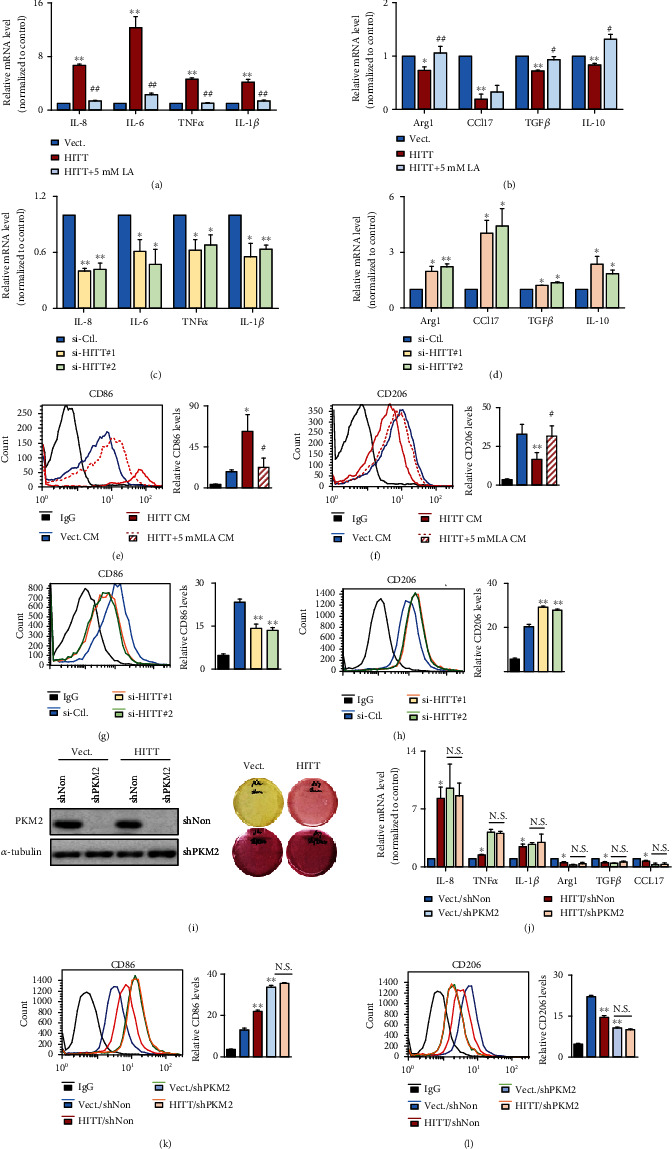
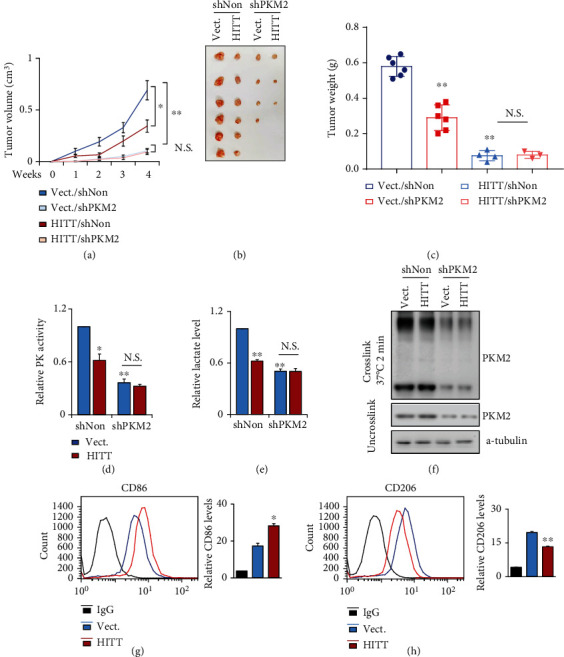
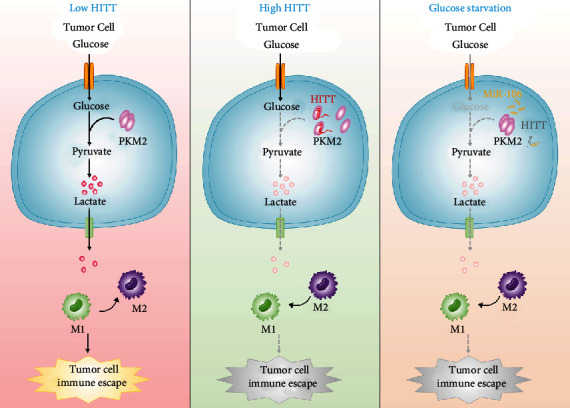
Lactic acid acidifies the tumor microenvironment and promotes multiple critical oncogenic processes, including immune evasion. Pyruvate kinase M2 (PKM2) is a dominant form of pyruvate kinase (PK) expressed in cancers that plays essential roles in metabolic reprograming and lactate production, rendering it as an attractive therapeutic target of cancer. However, the mechanism underlying PKM2 regulation remains unclear. Here, we show that long noncoding RNA (lncRNA) HIF-1α inhibitor at transcription level (HITT) inhibits lactate production in a PKM2-dependent manner. Mechanistically, it physically interacts with PKM2 mapped to a region that has been involved in both dimer (less-active) and tetramer (more-active) formation, inhibiting PKM2 oligomerization and leading to dramatic reduction of PK activity. Under glucose starvation, HITT was reduced as a result of miR-106 induction, which subsequently facilitates PKM2 oligomerization and increases vulnerability to apoptosis under glucose starvation stress. In addition, the interaction also reduces lactate secretion from cancer cells, which subsequently polarizes macrophages toward an M2-like anti-inflammatory phenotype and thus possibly contributes to immune escape in vivo. This study highlights an important role of an lncRNA in regulating PKM2 activity and also reveals a metabolic regulatory effect of PKM2 on macrophage polarization.
Copyright © 2022 Kunming Zhao et al.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
LinkOut - more resources
Full Text Sources
Miscellaneous
