

Genetic Diversity and Low Therapeutic Impact of Variant-Specific Markers in HIV-1 Pol Proteins
- PMID: 35910645
- PMCID: PMC9330395
- DOI: 10.3389/fmicb.2022.866705
Genetic Diversity and Low Therapeutic Impact of Variant-Specific Markers in HIV-1 Pol Proteins
Abstract
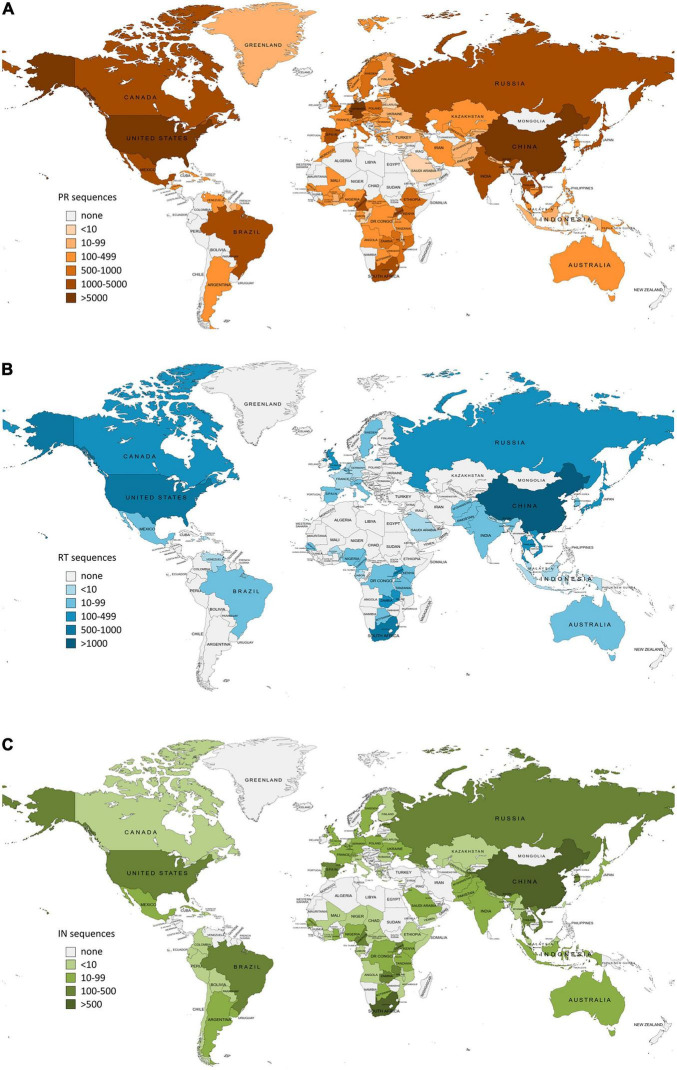
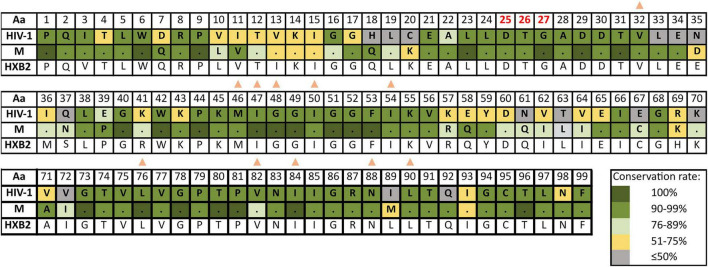
The emergence and spread of new HIV-1 variants pose a challenge for the effectiveness of antiretrovirals (ARV) targeting Pol proteins. During viral evolution, non-synonymous mutations have fixed along the viral genome, leading to amino acid (aa) changes that can be variant-specific (V-markers). Those V-markers fixed in positions associated with drug resistance mutations (DRM), or R-markers, can impact drug susceptibility and resistance pathways. All available HIV-1 Pol sequences from ARV-naïve subjects were downloaded from the United States Los Alamos HIV Sequence Database, selecting 59,733 protease (PR), 6,437 retrotranscriptase (RT), and 6,059 integrase (IN) complete sequences ascribed to the four HIV-1 groups and group M subtypes and circulating recombinant forms (CRFs). Using a bioinformatics tool developed in our laboratory (EpiMolBio), we inferred the consensus sequences for each Pol protein and HIV-1 variant to analyze the aa conservation in Pol. We analyzed the Wu-Kabat protein variability coefficient (WK) in PR, RT, and IN group M to study the susceptibility of each site to evolutionary replacements. We identified as V-markers the variant-specific aa changes present in >75% of the sequences in variants with >5 available sequences, considering R-markers those V-markers that corresponded to DRM according to the IAS-USA2019 and Stanford-Database 9.0. The mean aa conservation of HIV-1 and group M consensus was 82.60%/93.11% in PR, 88.81%/94.07% in RT, and 90.98%/96.02% in IN. The median group M WK was 10 in PR, 4 in RT, and 5 in IN. The residues involved in binding or catalytic sites showed a variability <0.5%. We identified 106 V-markers: 31 in PR, 28 in RT, and 47 in IN, present in 11, 12, and 13 variants, respectively. Among them, eight (7.5%) were R-markers, present in five variants, being minor DRM with little potential effect on ARV susceptibility. We present a thorough analysis of Pol variability among all HIV-1 variants circulating to date. The relatively high aa conservation observed in Pol proteins across HIV-1 variants highlights their critical role in the viral cycle. However, further studies are needed to understand the V-markers' impact on the Pol proteins structure, viral cycle, or treatment strategies, and periodic variability surveillance studies are also required to understand PR, RT, and IN evolution.
Keywords: HIV-1; Pol; conservation; integrase; protease; resistance; reverse transcriptase; variants.
Copyright © 2022 Troyano-Hernáez, Reinosa and Holguín.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures






References
-
- Baxter J. D., Schapiro J. M., Boucher C. A. B., Kohlbrenner V. M., Hall D. B., Scherer J. R., et al. (2006). Genotypic changes in human immunodeficiency virus type 1 protease associated with reduced susceptibility and virologic response to the protease inhibitor tipranavir. J. Virol. 80 10794–10801. 10.1128/JVI.00712-06 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials