

DCLK1 promotes colorectal cancer stemness and aggressiveness via the XRCC5/COX2 axis
- PMID: 35910805
- PMCID: PMC9330537
- DOI: 10.7150/thno.72037
DCLK1 promotes colorectal cancer stemness and aggressiveness via the XRCC5/COX2 axis
Abstract
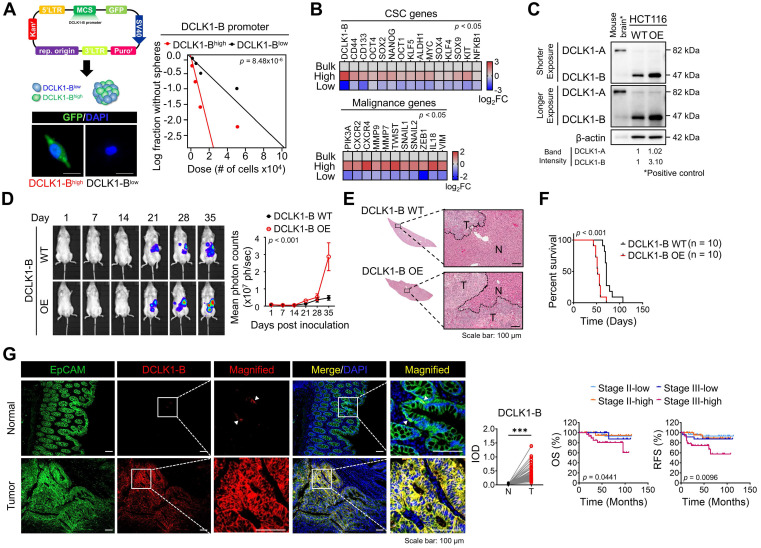
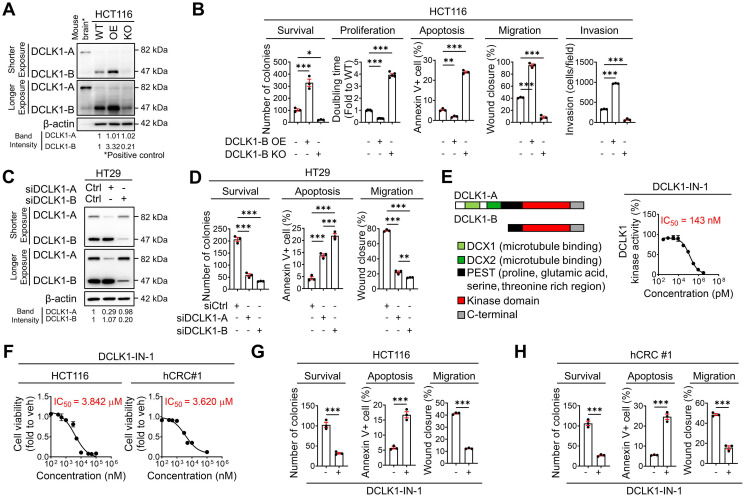
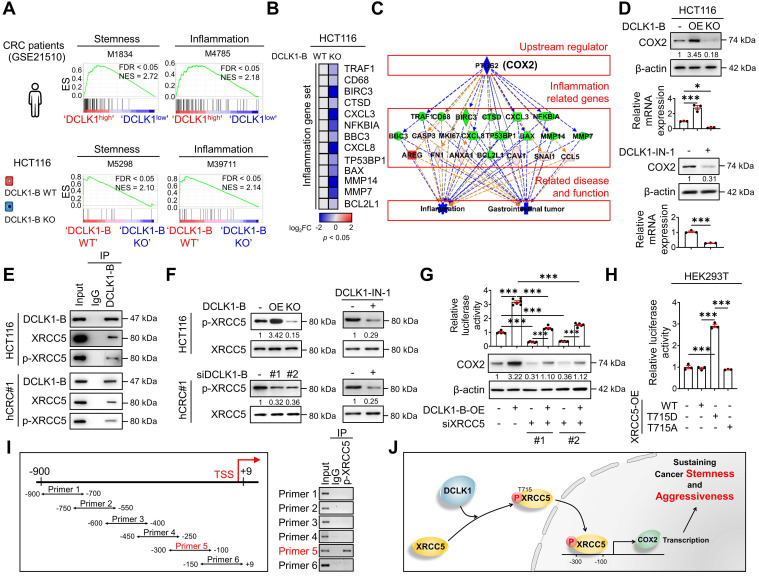
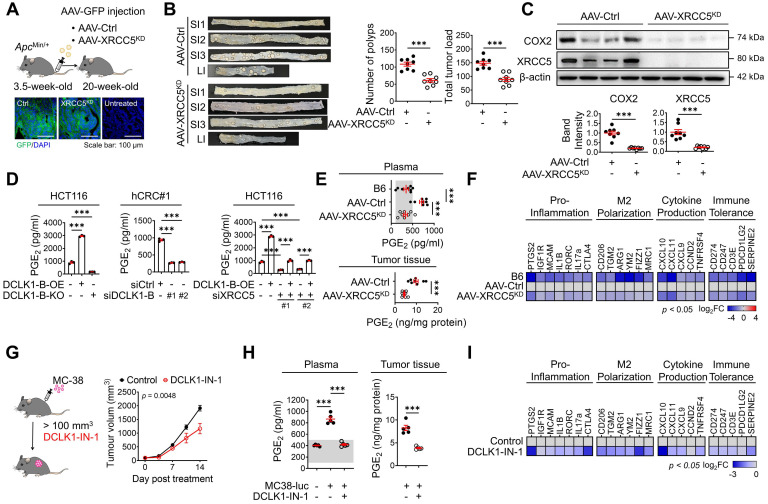
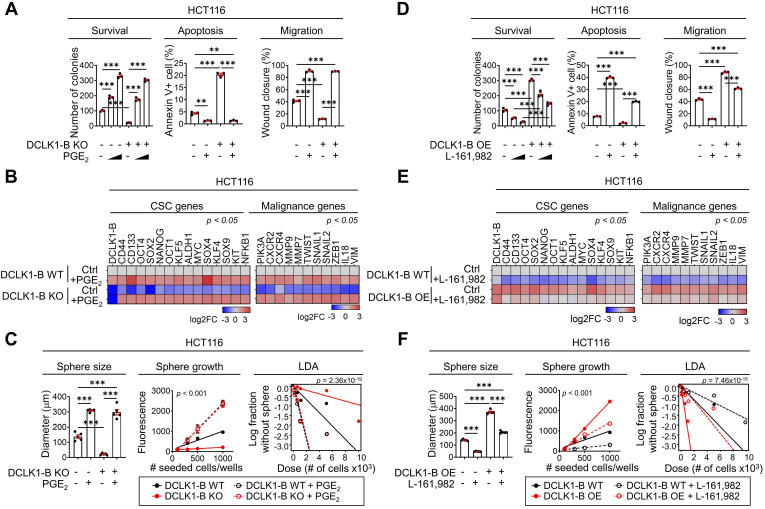
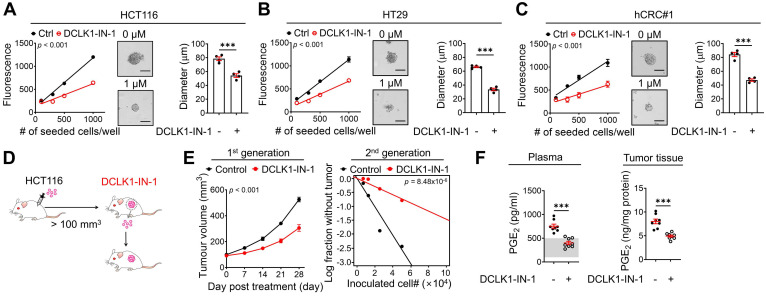
Rationale: Doublecortin-like kinase 1 (DCLK1) is a serine/threonine kinase that selectively marks cancer stem-like cells (CSCs) and promotes malignant progression in colorectal cancer (CRC). However, the exact molecular mechanism by which DCLK1 drives the aggressive phenotype of cancer cells is incompletely determined. Methods: Here, we performed comprehensive genomics and proteomics analyses to identify binding proteins of DCLK1 and discovered X-ray repair cross-complementing 5 (XRCC5). Thus, we explored the biological role and downstream events of the DCLK1/XRCC5 axis in human CRC cells and CRC mouse models. Results: The results of comprehensive bioinformatics analyses suggested that DCLK1-driven CRC aggressiveness is linked to inflammation. Mechanistically, DCLK1 bound and phosphorylated XRCC5, which in turn transcriptionally activated cyclooxygenase-2 expression and enhanced prostaglandin E2 production; these events collectively generated the inflammatory tumor microenvironment and enhanced the aggressive behavior of CRC cells. Consistent with the discovered mechanism, inhibition of DCLK1 kinase activity strongly impaired the tumor seeding and growth capabilities in CRC mouse models. Conclusion: Our study illuminates a novel mechanism that mediates the pro-inflammatory function of CSCs in driving the aggressive phenotype of CRC, broadening the biological function of DCLK1 in CRC.
Keywords: Cancer stem cells; Doublecortin-like kinase 1; Inflammatory tumor microenvironment; Prostaglandin E2.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
Similar articles
-
ELF1's Role in Colorectal Cancer: Up-Regulating DCLK1 Expression to Propel Malignant Progression and Stemness Features.Discov Med. 2025 Feb;37(193):315-325. doi: 10.24976/Discov.Med.202537193.25. Discov Med. 2025. PMID: 39973554
-
Dclk1, a tumor stem cell marker, regulates pro-survival signaling and self-renewal of intestinal tumor cells.Mol Cancer. 2017 Feb 1;16(1):30. doi: 10.1186/s12943-017-0594-y. Mol Cancer. 2017. Retraction in: Mol Cancer. 2025 Apr 19;24(1):120. doi: 10.1186/s12943-025-02329-3. PMID: 28148261 Free PMC article. Retracted.
-
Doublecortin-like kinase 1 promotes stem cell-like properties through the Hippo-YAP pathway in prostate cancer.Int J Med Sci. 2025 Jan 1;22(2):460-472. doi: 10.7150/ijms.99062. eCollection 2025. Int J Med Sci. 2025. PMID: 39781521 Free PMC article.
-
The Essential Role of DCLK1 in Pathogenesis, Diagnostic Procedures and Prognostic Stratification of Colorectal Cancer.Anticancer Res. 2019 Jun;39(6):2689-2697. doi: 10.21873/anticanres.13394. Anticancer Res. 2019. PMID: 31177103 Review.
-
DCLK1 in gastrointestinal cancer: A driver of tumor progression and a promising therapeutic target.Int J Cancer. 2025 Jun 1;156(11):2068-2086. doi: 10.1002/ijc.35365. Epub 2025 Mar 8. Int J Cancer. 2025. PMID: 40056091 Review.
Cited by
-
From Inflammation to Oncogenesis: Tracing Serum DCLK1 and miRNA Signatures in Chronic Liver Diseases.Int J Mol Sci. 2024 Jun 12;25(12):6481. doi: 10.3390/ijms25126481. Int J Mol Sci. 2024. PMID: 38928187 Free PMC article.
-
Role of DCLK1/Hippo pathway in type II alveolar epithelial cells differentiation in acute respiratory distress syndrome.Mol Med. 2023 Nov 23;29(1):159. doi: 10.1186/s10020-023-00760-0. Mol Med. 2023. PMID: 37996782 Free PMC article.
-
Expression of Stem Cell Markers in High-LET Space Radiation-Induced Intestinal Tumors in Apc1638N/+ Mouse Intestine.Cancers (Basel). 2023 Aug 24;15(17):4240. doi: 10.3390/cancers15174240. Cancers (Basel). 2023. PMID: 37686516 Free PMC article.
-
Epigenetic Landscape and Therapeutic Implication of Gene Isoforms of Doublecortin-Like Kinase 1 for Cancer Stem Cells.Int J Mol Sci. 2023 Nov 16;24(22):16407. doi: 10.3390/ijms242216407. Int J Mol Sci. 2023. PMID: 38003596 Free PMC article. Review.
-
X-ray cross-complementing family: the bridge linking DNA damage repair and cancer.J Transl Med. 2023 Sep 7;21(1):602. doi: 10.1186/s12967-023-04447-2. J Transl Med. 2023. PMID: 37679817 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
