

4-Phenylbutyrate restored γ-aminobutyric acid uptake and reduced seizures in SLC6A1 patient variant-bearing cell and mouse models
- PMID: 35911425
- PMCID: PMC9336585
- DOI: 10.1093/braincomms/fcac144
4-Phenylbutyrate restored γ-aminobutyric acid uptake and reduced seizures in SLC6A1 patient variant-bearing cell and mouse models
Abstract
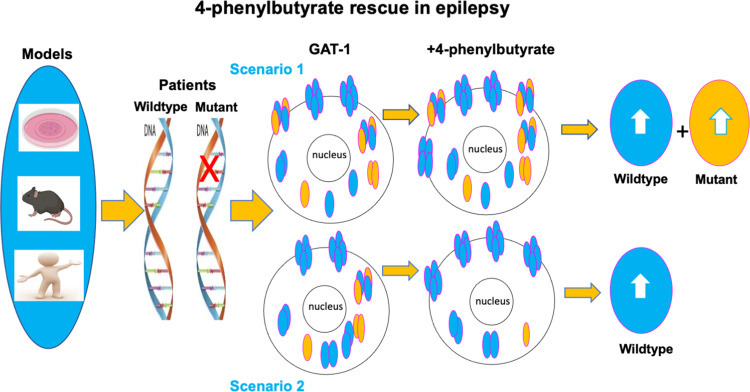
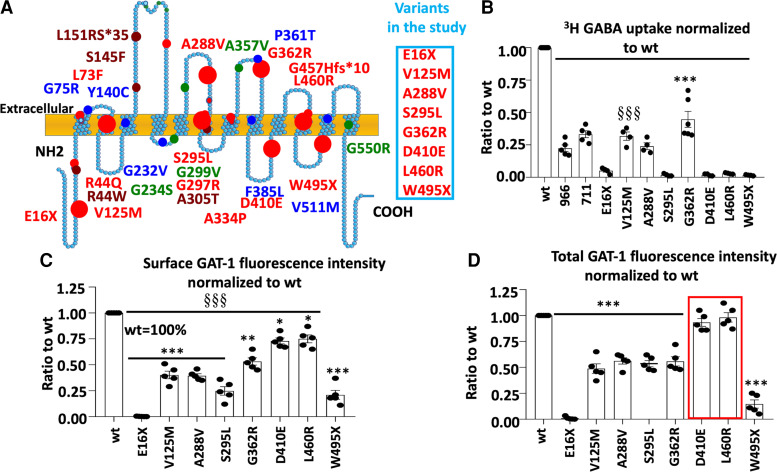
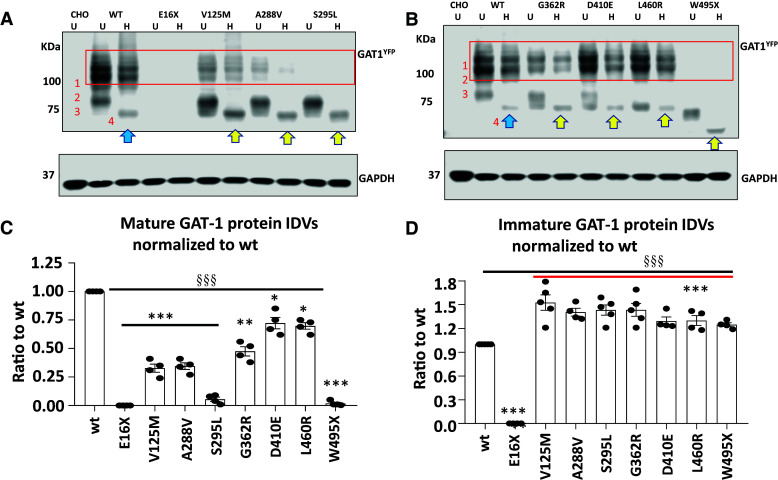
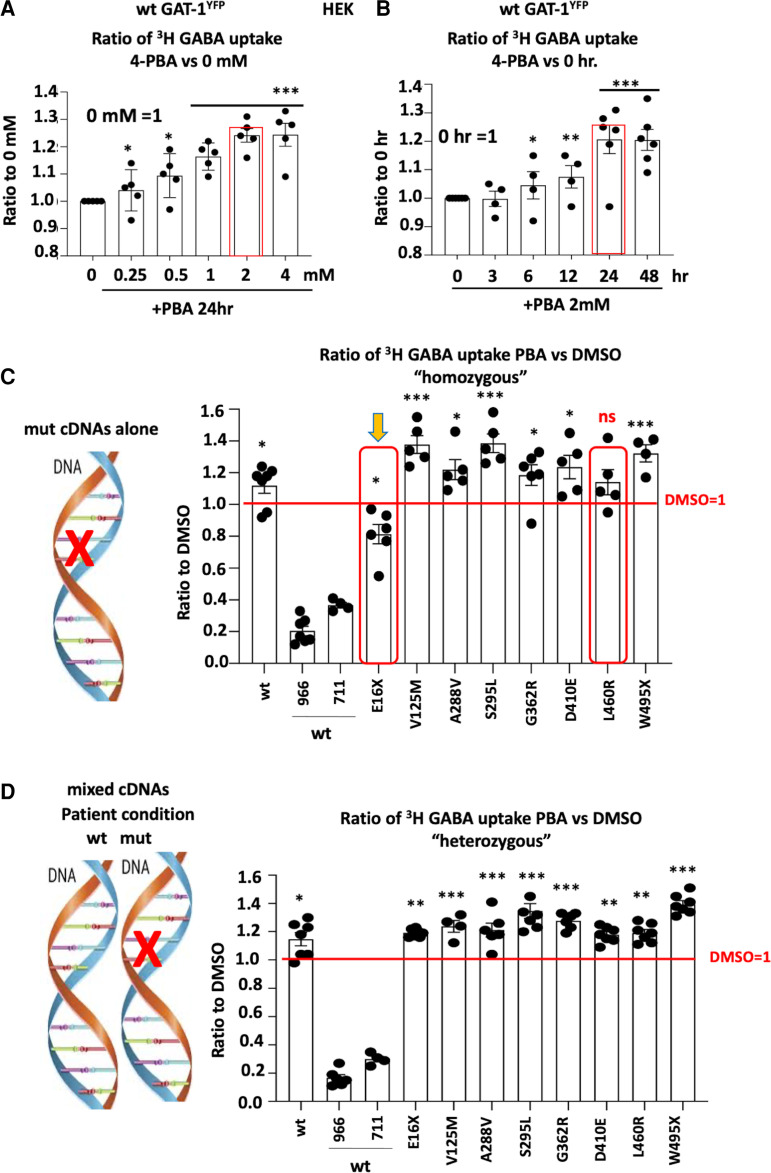
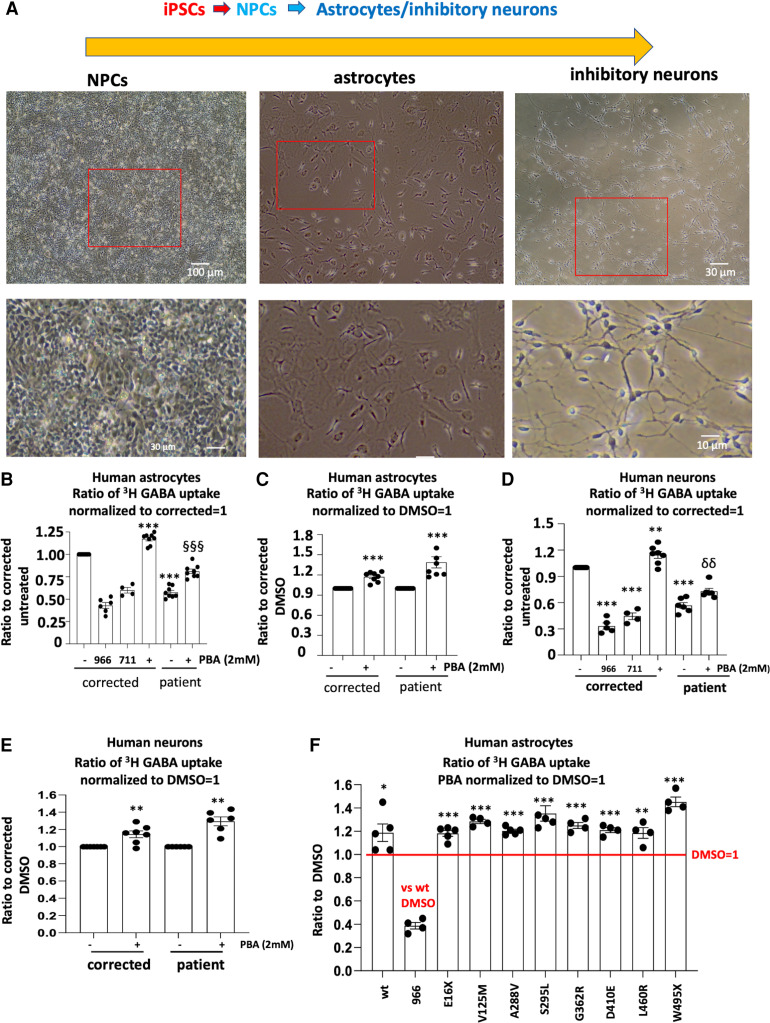
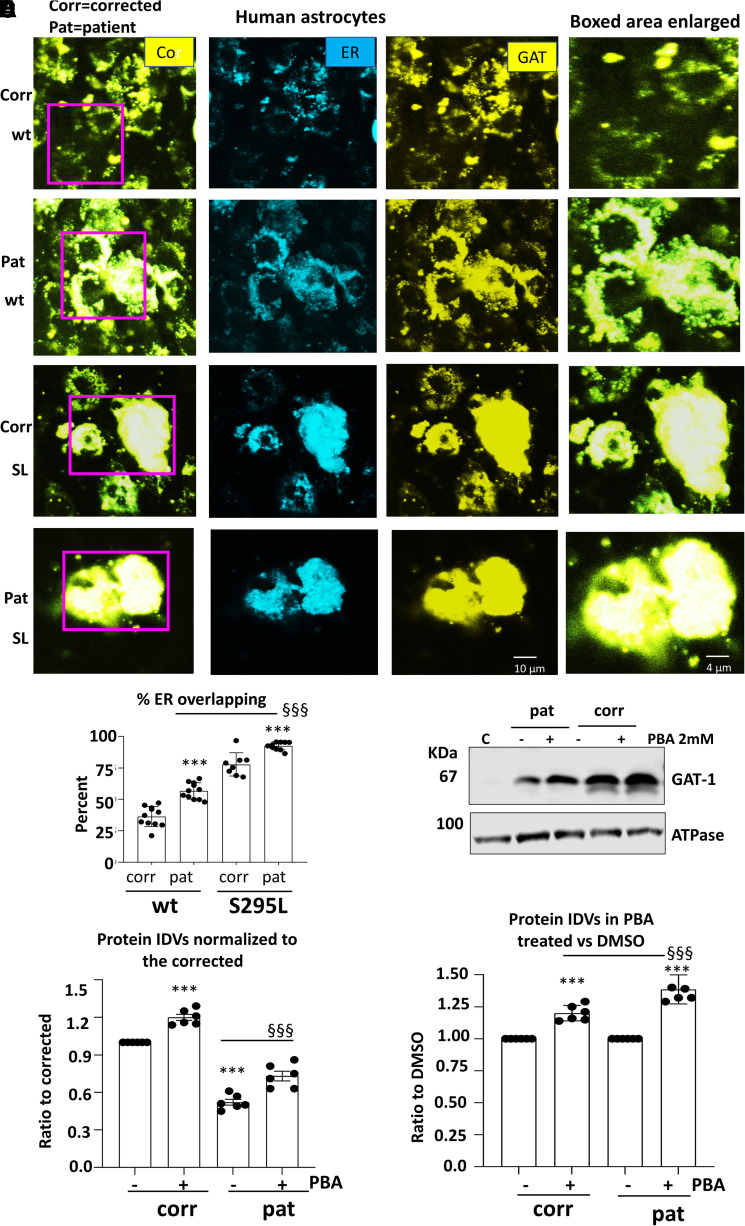
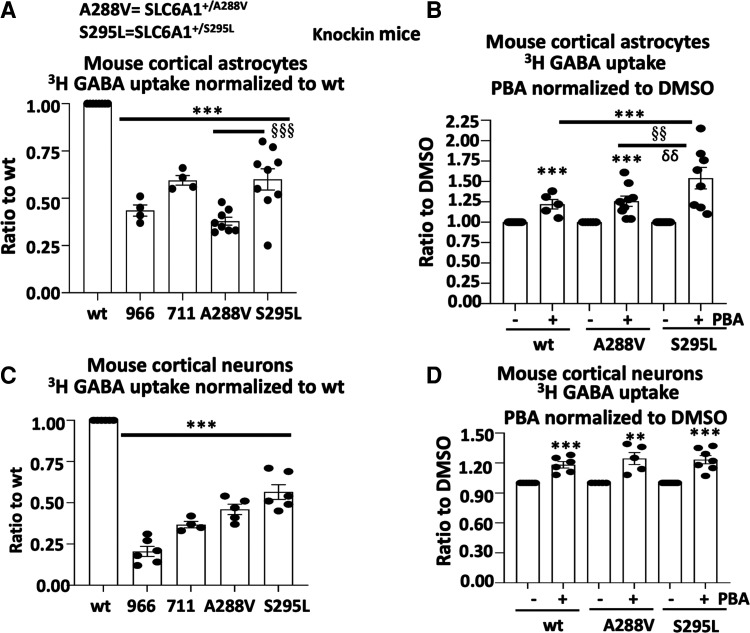
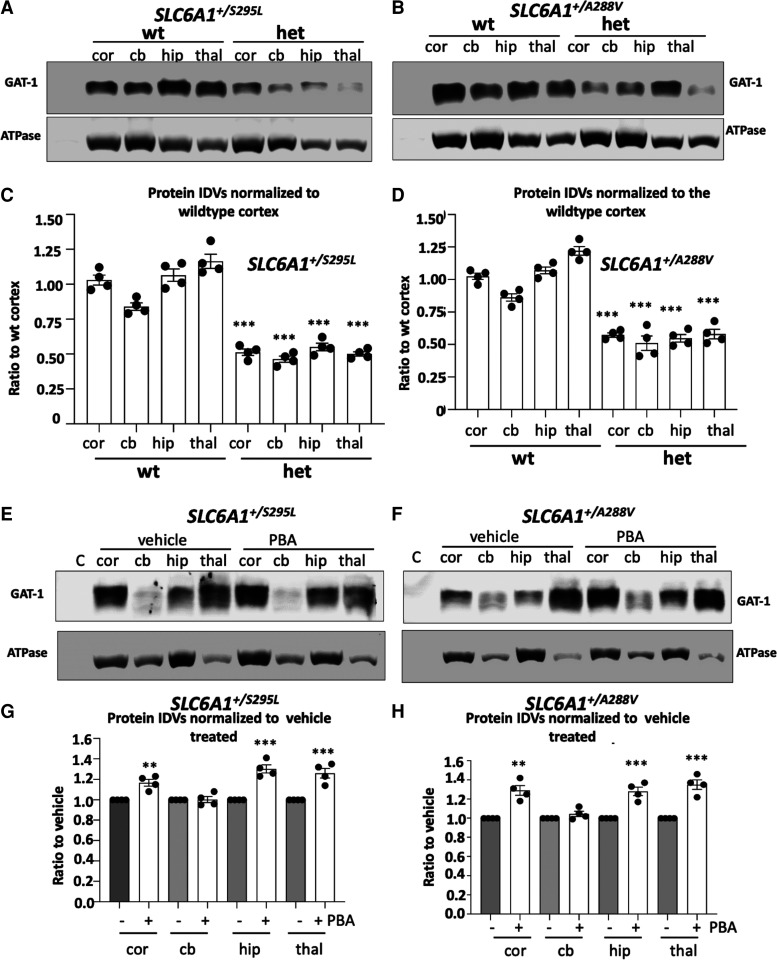
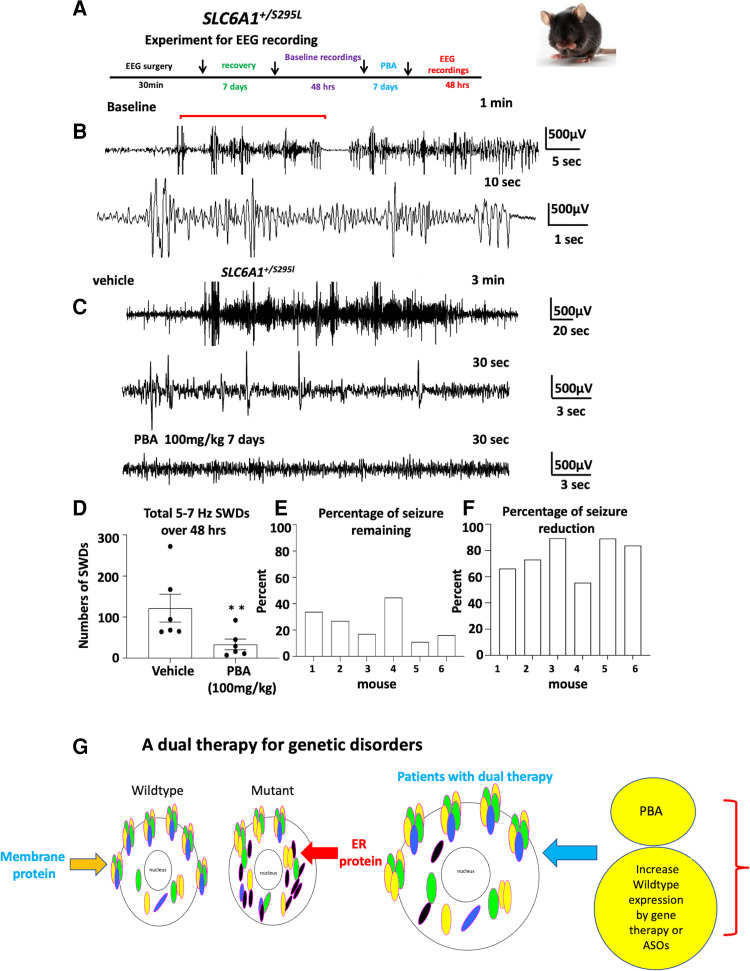
We have studied the molecular mechanisms of variants in solute carrier Family 6 Member 1 associated with neurodevelopmental disorders, including various epilepsy syndromes, autism and intellectual disability. Based on functional assays of solute carrier Family 6 Member 1 variants, we conclude that partial or complete loss of γ-amino butyric acid uptake due to reduced membrane γ-amino butyric acid transporter 1 trafficking is the primary aetiology. Importantly, we identified common patterns of the mutant γ-amino butyric acid transporter 1 protein trafficking from biogenesis, oligomerization, glycosylation and translocation to the cell membrane across variants in different cell types such as astrocytes and neurons. We hypothesize that therapeutic approaches to facilitate membrane trafficking would increase γ-amino butyric acid transporter 1 protein membrane expression and function. 4-Phenylbutyrate is a Food and Drug Administration-approved drug for paediatric use and is orally bioavailable. 4-Phenylbutyrate shows promise in the treatment of cystic fibrosis. The common cellular mechanisms shared by the mutant γ-amino butyric acid transporter 1 and cystic fibrosis transmembrane conductance regulator led us to hypothesize that 4-phenylbutyrate could be a potential treatment option for solute carrier Family 6 Member 1 mutations. We examined the impact of 4-phenylbutyrate across a library of variants in cell and knockin mouse models. Because γ-amino butyric acid transporter 1 is expressed in both neurons and astrocytes, and γ-amino butyric acid transporter 1 deficiency in astrocytes has been hypothesized to underlie seizure generation, we tested the effect of 4-phenylbutyrate in both neurons and astrocytes with a focus on astrocytes. We demonstrated existence of the mutant γ-amino butyric acid transporter 1 retaining wildtype γ-amino butyric acid transporter 1, suggesting the mutant protein causes aberrant protein oligomerization and trafficking. 4-Phenylbutyrate increased γ-amino butyric acid uptake in both mouse and human astrocytes and neurons bearing the variants. Importantly, 4-phenylbutyrate alone increased γ-amino butyric acid transporter 1 expression and suppressed spike wave discharges in heterozygous knockin mice. Although the mechanisms of action for 4-phenylbutyrate are still unclear, with multiple possibly being involved, it is likely that 4-phenylbutyrate can facilitate the forward trafficking of the wildtype γ-amino butyric acid transporter 1 regardless of rescuing the mutant γ-amino butyric acid transporter 1, thus increasing γ-amino butyric acid uptake. All patients with solute carrier Family 6 Member 1 variants are heterozygous and carry one wildtype allele, suggesting a great opportunity for treatment development leveraging wildtype protein trafficking. The study opens a novel avenue of treatment development for genetic epilepsy via drug repurposing.
Keywords: 4-phenylbutyrate acid; GABA transporter 1 (GAT-1); autism; chaperone; epilepsy.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Guarantors of Brain.
Figures
Similar articles
-
Common molecular mechanisms of SLC6A1 variant-mediated neurodevelopmental disorders in astrocytes and neurons.Brain. 2021 Sep 4;144(8):2499-2512. doi: 10.1093/brain/awab207. Brain. 2021. PMID: 34028503 Free PMC article.
-
γ-Aminobutyric acid transporter and GABAA receptor mechanisms in Slc6a1+/A288V and Slc6a1+/S295L mice associated with developmental and epileptic encephalopathies.Brain Commun. 2024 Apr 22;6(2):fcae110. doi: 10.1093/braincomms/fcae110. eCollection 2024. Brain Commun. 2024. PMID: 38650830 Free PMC article.
-
Astrocytic GABA transporter 1 deficit in novel SLC6A1 variants mediated epilepsy: Connected from protein destabilization to seizures in mice and humans.Neurobiol Dis. 2022 Oct 1;172:105810. doi: 10.1016/j.nbd.2022.105810. Epub 2022 Jul 14. Neurobiol Dis. 2022. PMID: 35840120 Free PMC article.
-
Molecular and Clinical Repercussions of GABA Transporter 1 Variants Gone Amiss: Links to Epilepsy and Developmental Spectrum Disorders.Front Mol Biosci. 2022 Mar 2;9:834498. doi: 10.3389/fmolb.2022.834498. eCollection 2022. Front Mol Biosci. 2022. PMID: 35295842 Free PMC article. Review.
-
A transporter's doom or destiny: SLC6A1 in health and disease, novel molecular targets and emerging therapeutic prospects.Front Mol Neurosci. 2024 Aug 29;17:1466694. doi: 10.3389/fnmol.2024.1466694. eCollection 2024. Front Mol Neurosci. 2024. PMID: 39268250 Free PMC article. Review.
Cited by
-
Experimental and Bioinformatic Insights into the Effects of Epileptogenic Variants on the Function and Trafficking of the GABA Transporter GAT-1.Int J Mol Sci. 2023 Jan 4;24(2):955. doi: 10.3390/ijms24020955. Int J Mol Sci. 2023. PMID: 36674476 Free PMC article.
-
Identification of New Ciliary Signaling Pathways in the Brain and Insights into Neurological Disorders.J Neurosci. 2025 Aug 13;45(33):e0800242025. doi: 10.1523/JNEUROSCI.0800-24.2025. J Neurosci. 2025. PMID: 40623838 Free PMC article.
-
Patient-derived SLC6A1 variant S295L results in an epileptic phenotype similar to haploinsufficient mice.Epilepsia. 2023 Oct;64(10):e214-e221. doi: 10.1111/epi.17731. Epub 2023 Aug 8. Epilepsia. 2023. PMID: 37501613 Free PMC article.
-
Merritt-Putnam Symposium | Developmental and Epileptic Encephalopathies-Current Concepts and Novel Approaches.Epilepsy Curr. 2025 Mar 25;25(3):229-235. doi: 10.1177/15357597251320142. eCollection 2025 May-Jun. Epilepsy Curr. 2025. PMID: 40161506 Free PMC article. Review.
-
The phenotypic presentation of adult individuals with SLC6A1-related neurodevelopmental disorders.Front Neurosci. 2023 Aug 17;17:1216653. doi: 10.3389/fnins.2023.1216653. eCollection 2023. Front Neurosci. 2023. PMID: 37662110 Free PMC article.
References
-
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:S10–S17. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources