

Therapeutic potential of inhibiting histone 3 lysine 27 demethylases: a review of the literature
- PMID: 35915507
- PMCID: PMC9344682
- DOI: 10.1186/s13148-022-01305-8
Therapeutic potential of inhibiting histone 3 lysine 27 demethylases: a review of the literature
Abstract
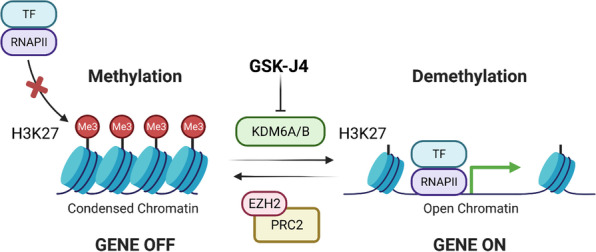
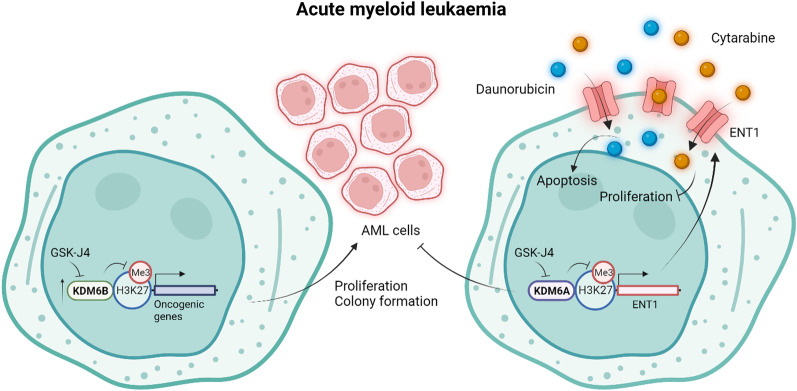
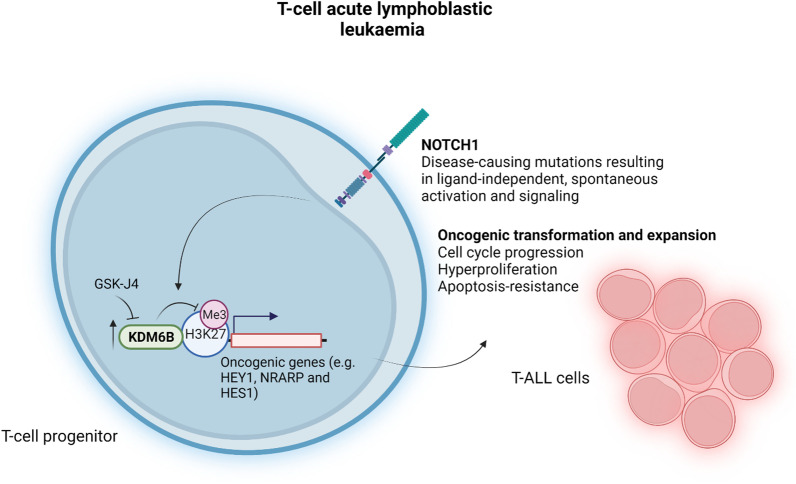
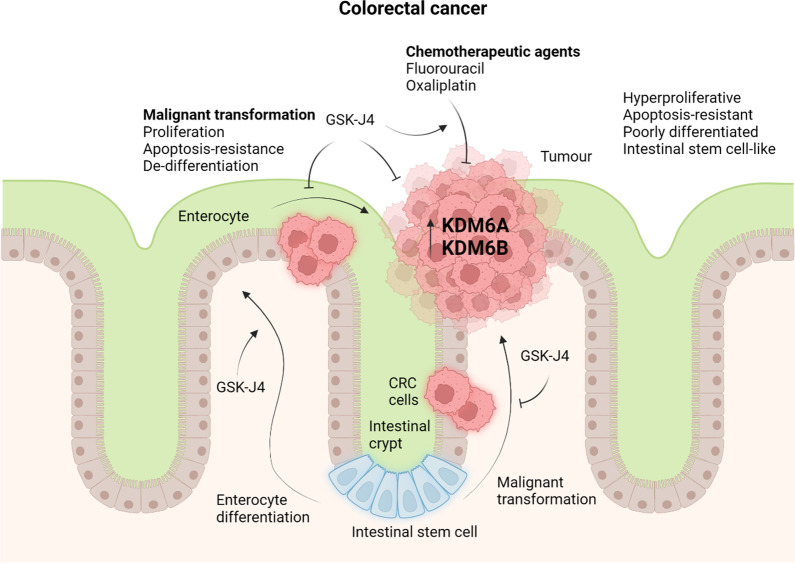
Histone 3 lysine 27 (H3K27) demethylation constitutes an important epigenetic mechanism of gene activation. It is mediated by the Jumonji C domain-containing lysine demethylases KDM6A and KDM6B, both of which have been implicated in a wide myriad of diseases, including blood and solid tumours, autoimmune and inflammatory disorders, and infectious diseases. Here, we review and summarise the pre-clinical evidence, both in vitro and in vivo, in support of the therapeutic potential of inhibiting H3K27-targeting demethylases, with a focus on the small-molecule inhibitor GSK-J4. In malignancies, KDM6A/B inhibition possesses the ability to inhibit proliferation, induce apoptosis, promote differentiation, and heighten sensitivity to currently employed chemotherapeutics. KDM6A/B inhibition also comprises a potent anti-inflammatory approach in inflammatory and autoimmune disorders associated with inappropriately exuberant inflammatory and autoimmune responses, restoring immunological homeostasis to inflamed tissues. With respect to infectious diseases, KDM6A/B inhibition can suppress the growth of infectious pathogens and attenuate the immunopathology precipitated by these pathogens. The pre-clinical in vitro and in vivo data, summarised in this review, suggest that inhibiting H3K27 demethylases holds immense therapeutic potential in many diseases.
Keywords: Autoimmune diseases; Cancer; Epigenetics; GSK-J4; H3K27; Histone lysine demethylase; Infectious diseases; Inflammation; JMJD3; KDM6A; KDM6B; UTX.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
JMJD3 as an epigenetic regulator in development and disease.Int J Biochem Cell Biol. 2015 Oct;67:148-57. doi: 10.1016/j.biocel.2015.07.006. Epub 2015 Jul 17. Int J Biochem Cell Biol. 2015. PMID: 26193001 Free PMC article. Review.
-
Lysine Demethylase KDM6A in Differentiation, Development, and Cancer.Mol Cell Biol. 2020 Sep 28;40(20):e00341-20. doi: 10.1128/MCB.00341-20. Print 2020 Sep 28. Mol Cell Biol. 2020. PMID: 32817139 Free PMC article. Review.
-
EZH2, JMJD3, and UTX epigenetically regulate hepatic plasticity inducing retro-differentiation and proliferation of liver cells.Cell Death Dis. 2019 Jul 8;10(7):518. doi: 10.1038/s41419-019-1755-2. Cell Death Dis. 2019. PMID: 31285428 Free PMC article.
-
Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases.Proc Natl Acad Sci U S A. 2007 Nov 20;104(47):18439-44. doi: 10.1073/pnas.0707292104. Epub 2007 Nov 14. Proc Natl Acad Sci U S A. 2007. PMID: 18003914 Free PMC article.
-
Pharmacological targeting of KDM6A and KDM6B, as a novel therapeutic strategy for treating craniosynostosis in Saethre-Chotzen syndrome.Stem Cell Res Ther. 2020 Dec 9;11(1):529. doi: 10.1186/s13287-020-02051-5. Stem Cell Res Ther. 2020. PMID: 33298158 Free PMC article.
Cited by
-
Sex-specific epigenetic programming in renal fibrosis and inflammation.Am J Physiol Renal Physiol. 2023 Nov 1;325(5):F578-F594. doi: 10.1152/ajprenal.00091.2023. Epub 2023 Aug 10. Am J Physiol Renal Physiol. 2023. PMID: 37560775 Free PMC article. Review.
-
An Epigenetic Manifestation of Alzheimer's Disease: DNA Methylation.Actas Esp Psiquiatr. 2024 Jun;52(3):365-374. doi: 10.62641/aep.v52i3.1595. Actas Esp Psiquiatr. 2024. PMID: 38863055 Free PMC article. Review.
-
Integrative bioinformatics and validation studies reveal KDM6B and its associated molecules as crucial modulators in Idiopathic Pulmonary Fibrosis.Front Immunol. 2023 May 19;14:1183871. doi: 10.3389/fimmu.2023.1183871. eCollection 2023. Front Immunol. 2023. PMID: 37275887 Free PMC article.
-
MiR-26b-5p/TET3 regulates the osteogenic differentiation of human bone mesenchymal stem cells and bone reconstruction in female rats with calvarial defects.Mol Biol Rep. 2024 May 9;51(1):632. doi: 10.1007/s11033-024-09577-4. Mol Biol Rep. 2024. PMID: 38724827
-
Genome-wide CRISPR/Cas9 screening identifies PTGR2 as a potential therapeutic target for sunitinib resistance in clear cell renal cell carcinoma.Sci Rep. 2025 Jul 19;15(1):26263. doi: 10.1038/s41598-025-12192-3. Sci Rep. 2025. PMID: 40683967 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
