

Evaluating the impact of X-ray damage on conformational heterogeneity in room-temperature (277 K) and cryo-cooled protein crystals
- PMID: 35916220
- PMCID: PMC9344472
- DOI: 10.1107/S2059798322005939
Evaluating the impact of X-ray damage on conformational heterogeneity in room-temperature (277 K) and cryo-cooled protein crystals
Abstract
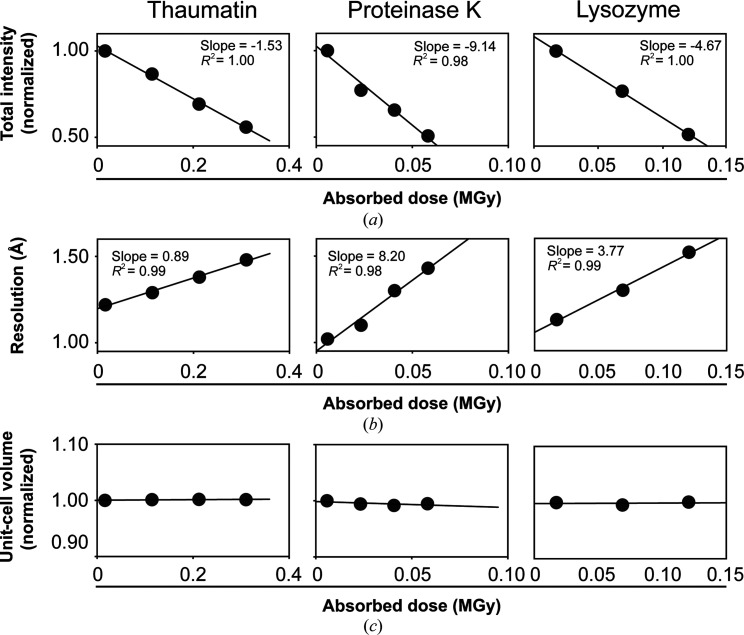
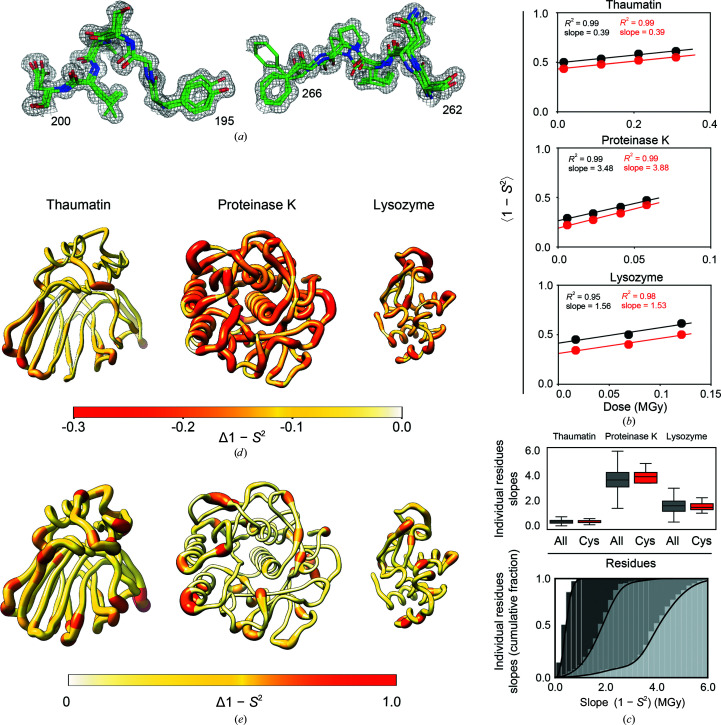
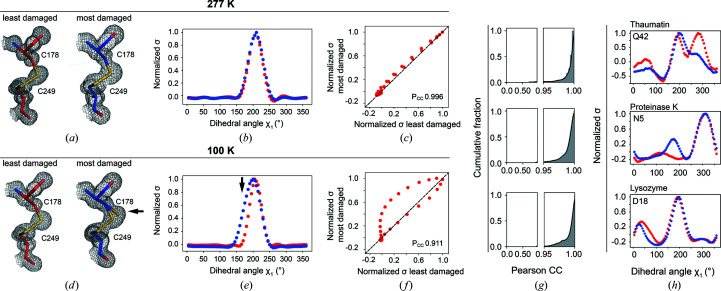
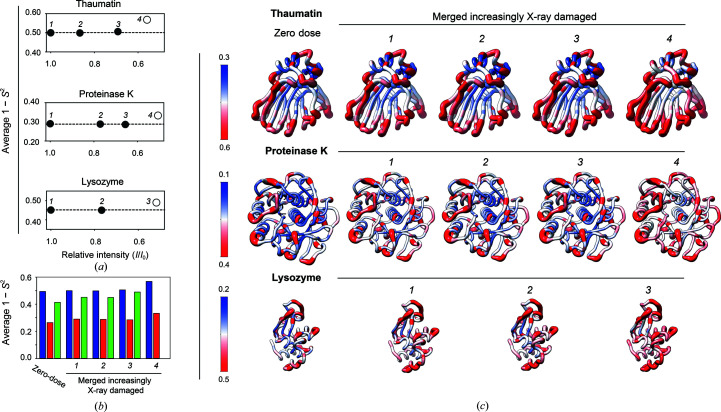
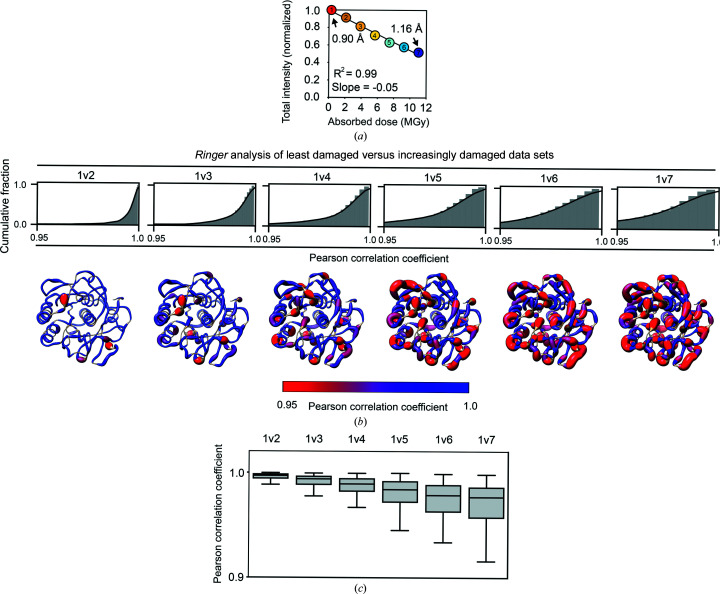
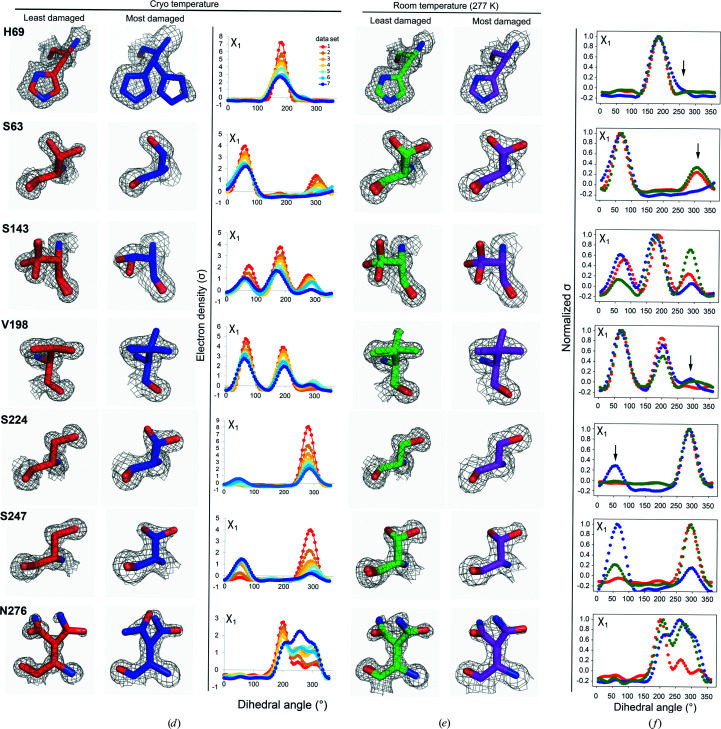
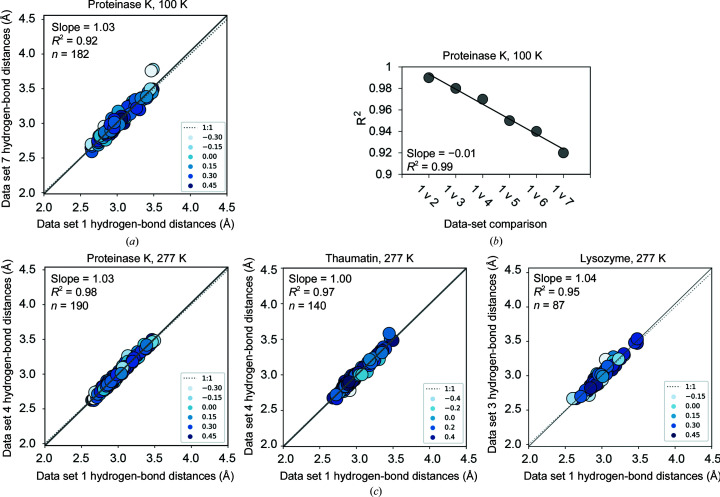
Cryo-cooling has been nearly universally adopted to mitigate X-ray damage and facilitate crystal handling in protein X-ray crystallography. However, cryo X-ray crystallographic data provide an incomplete window into the ensemble of conformations that is at the heart of protein function and energetics. Room-temperature (RT) X-ray crystallography provides accurate ensemble information, and recent developments allow conformational heterogeneity (the experimental manifestation of ensembles) to be extracted from single-crystal data. Nevertheless, high sensitivity to X-ray damage at RT raises concerns about data reliability. To systematically address this critical issue, increasingly X-ray-damaged high-resolution data sets (1.02-1.52 Å resolution) were obtained from single proteinase K, thaumatin and lysozyme crystals at RT (277 K). In each case a modest increase in conformational heterogeneity with X-ray damage was observed. Merging data with different extents of damage (as is typically carried out) had negligible effects on conformational heterogeneity until the overall diffraction intensity decayed to ∼70% of its initial value. These effects were compared with X-ray damage effects in cryo-cooled crystals by carrying out an analogous analysis of increasingly damaged proteinase K cryo data sets (0.9-1.16 Å resolution). X-ray damage-associated heterogeneity changes were found that were not observed at RT. This property renders it difficult to distinguish real from artefactual conformations and to determine the conformational response to changes in temperature. The ability to acquire reliable heterogeneity information from single crystals at RT, together with recent advances in RT data collection at accessible synchrotron beamlines, provides a strong motivation for the widespread adoption of RT X-ray crystallography to obtain conformational ensemble information.
Keywords: X-ray damage; conformational ensembles; hydrogen bonds; protein X-ray crystallography; room temperature.
Figures
References
-
- Adam, V., Carpentier, P., Violot, S., Lelimousin, M., Darnault, C., Nienhaus, G. U. & Bourgeois, D. (2009). J. Am. Chem. Soc. 131, 18063–18065. - PubMed
-
- Alford, R. F., Leaver-Fay, A., Jeliazkov, J. R., O’Meara, M. J., DiMaio, F. P., Park, H., Shapovalov, M. V., Renfrew, P. D., Mulligan, V. K., Kappel, K., Labonte, J. W., Pacella, M. S., Bonneau, R., Bradley, P., Dunbrack, R. L., Das, R., Baker, D., Kuhlman, B., Kortemme, T. & Gray, J. J. (2017). J. Chem. Theory Comput. 13, 3031–3048. - PMC - PubMed
-
- Austin, R. H., Beeson, K. W., Eisenstein, L., Frauenfelder, H. & Gunsalus, I. C. (1975). Biochemistry, 14, 5355–5373. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
