

Selenium Modulates Cancer Cell Response to Pharmacologic Ascorbate
- PMID: 35916672
- PMCID: PMC9532358
- DOI: 10.1158/0008-5472.CAN-22-0408
Selenium Modulates Cancer Cell Response to Pharmacologic Ascorbate
Abstract
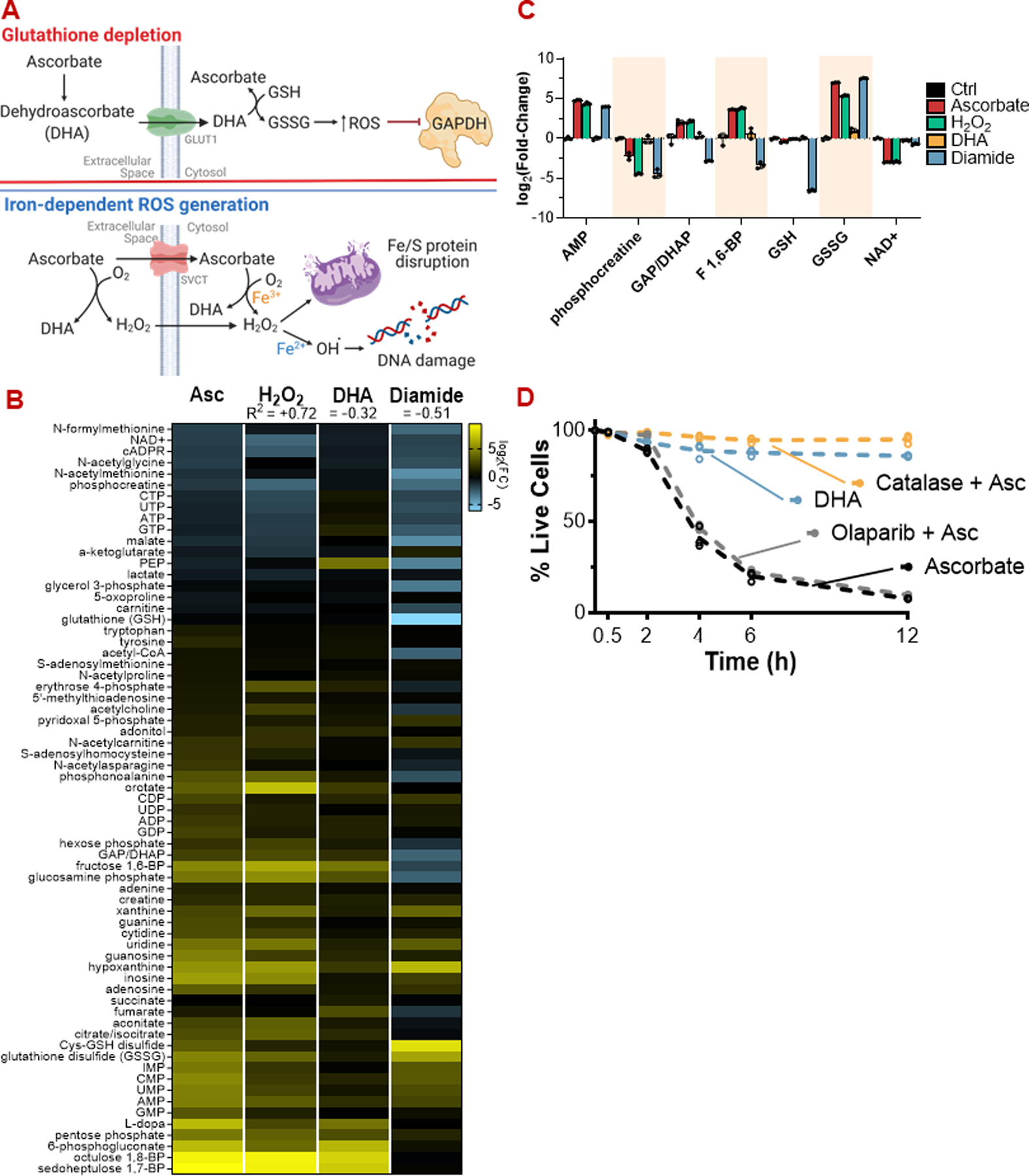
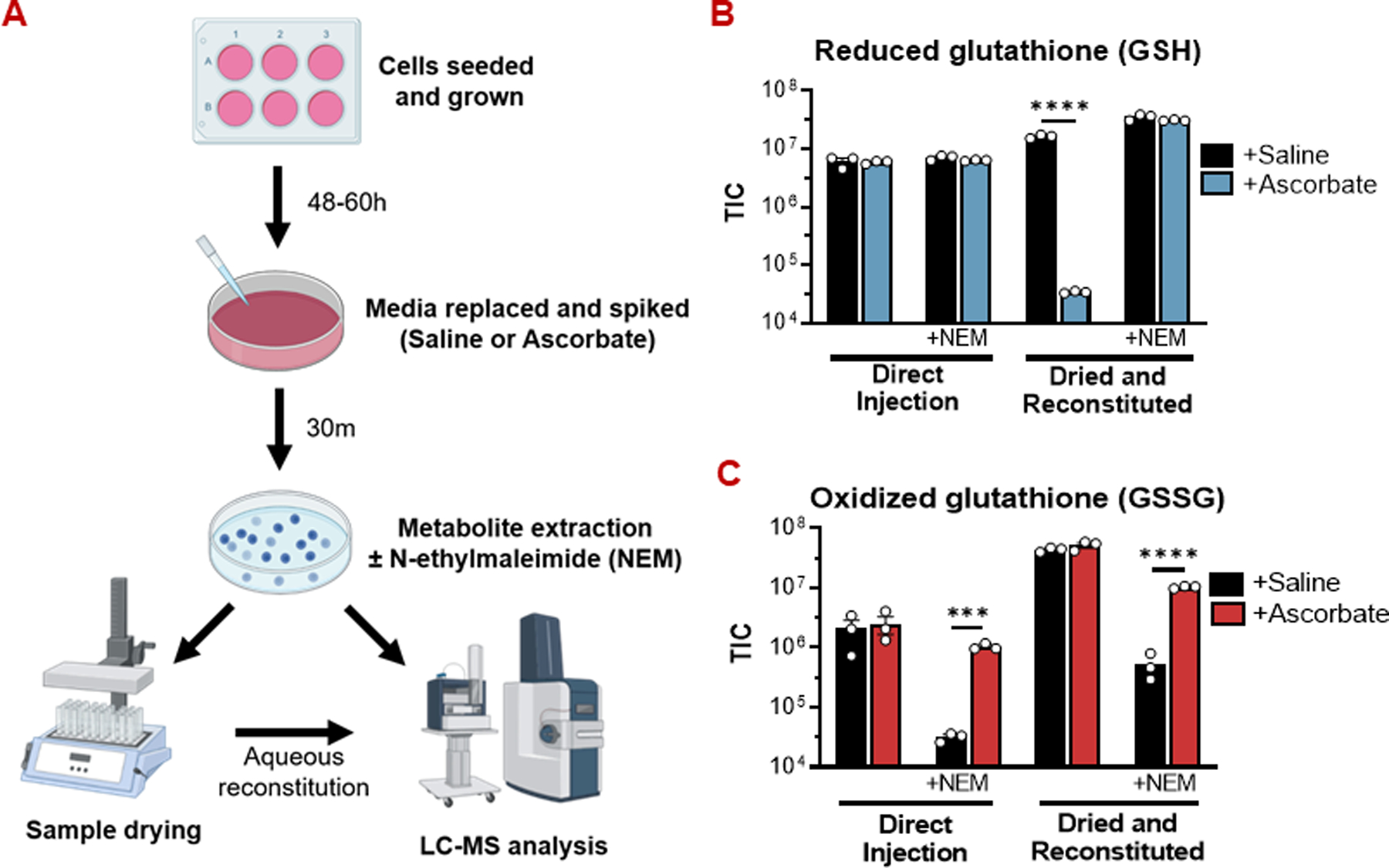
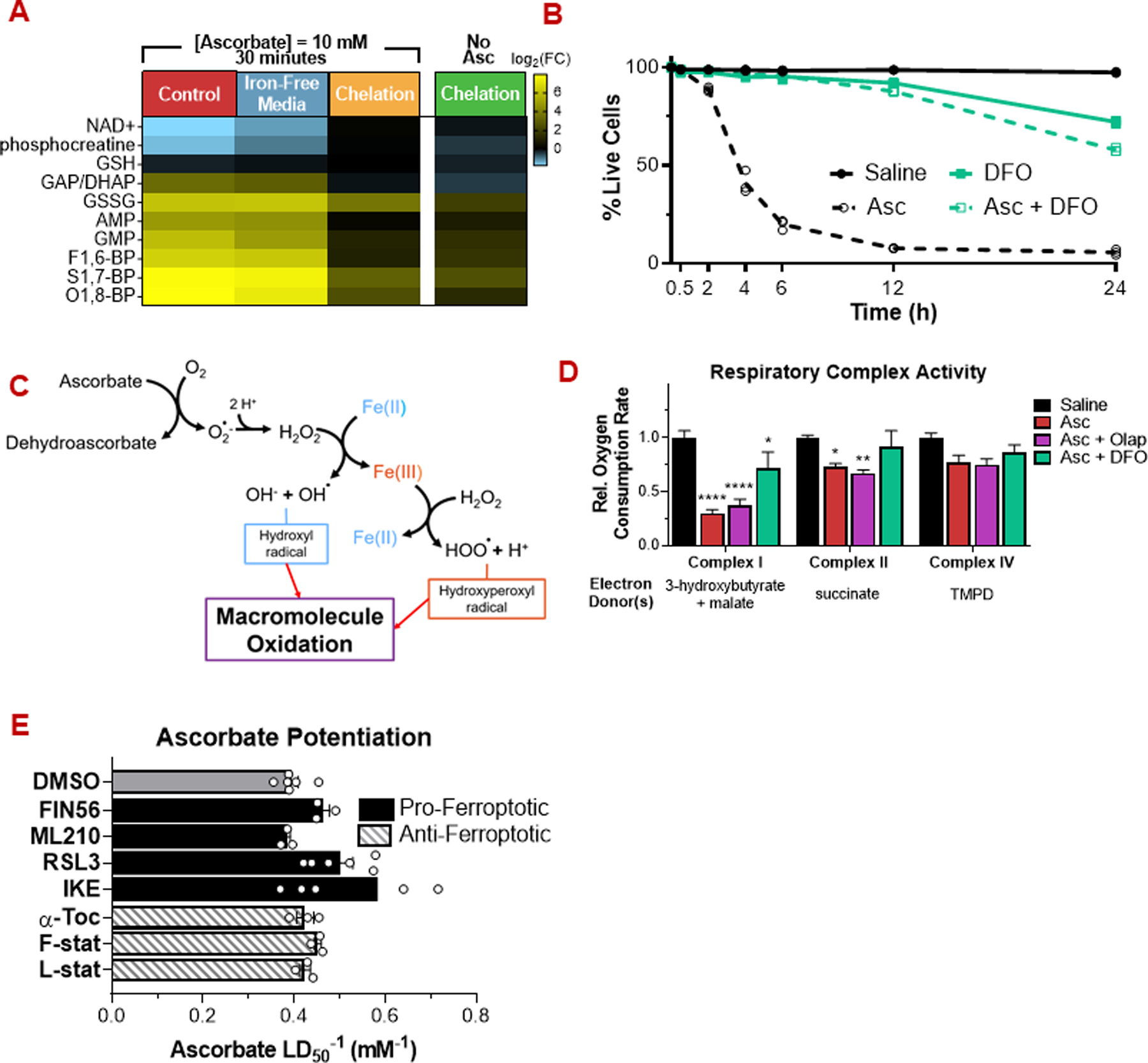
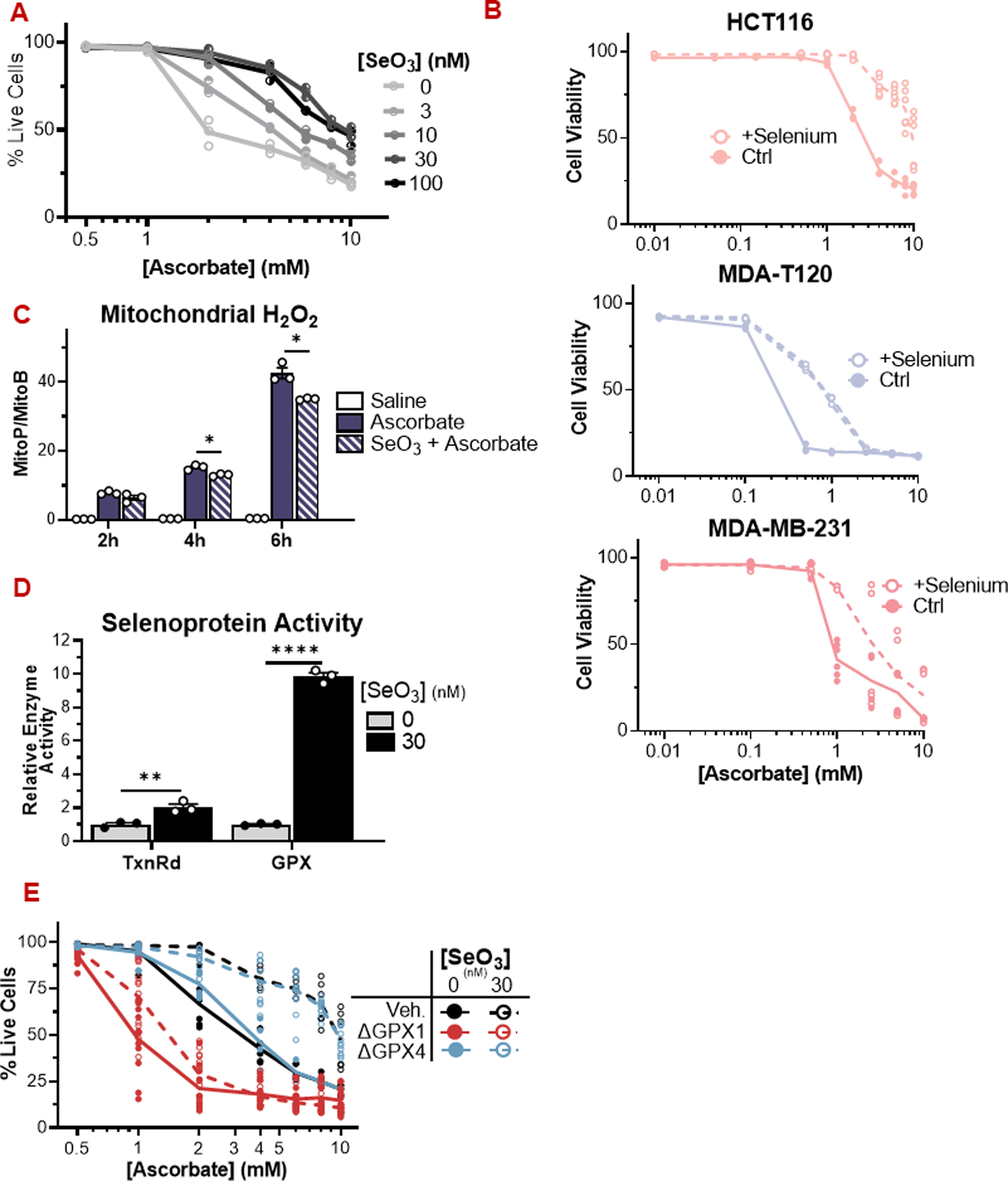
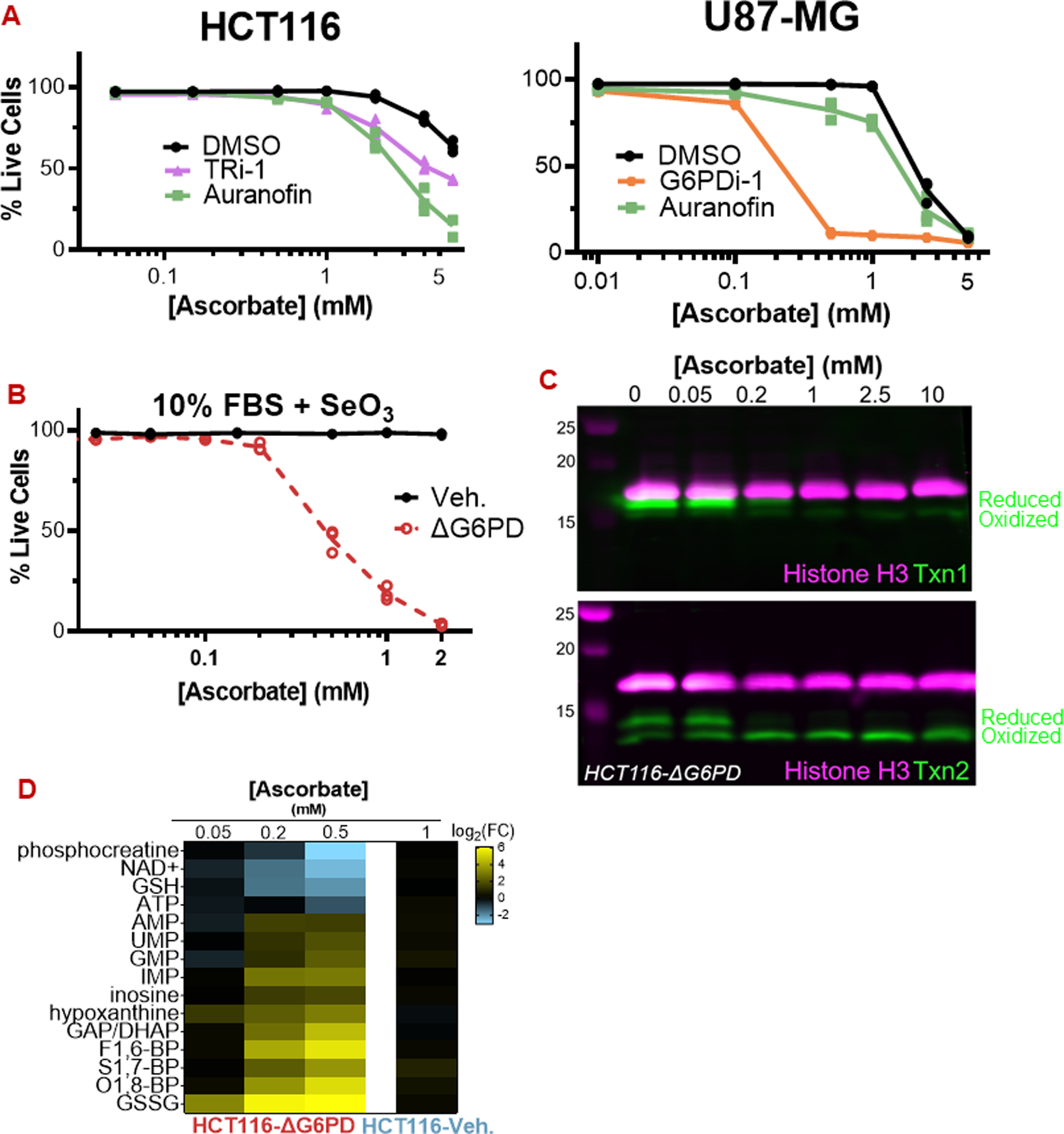
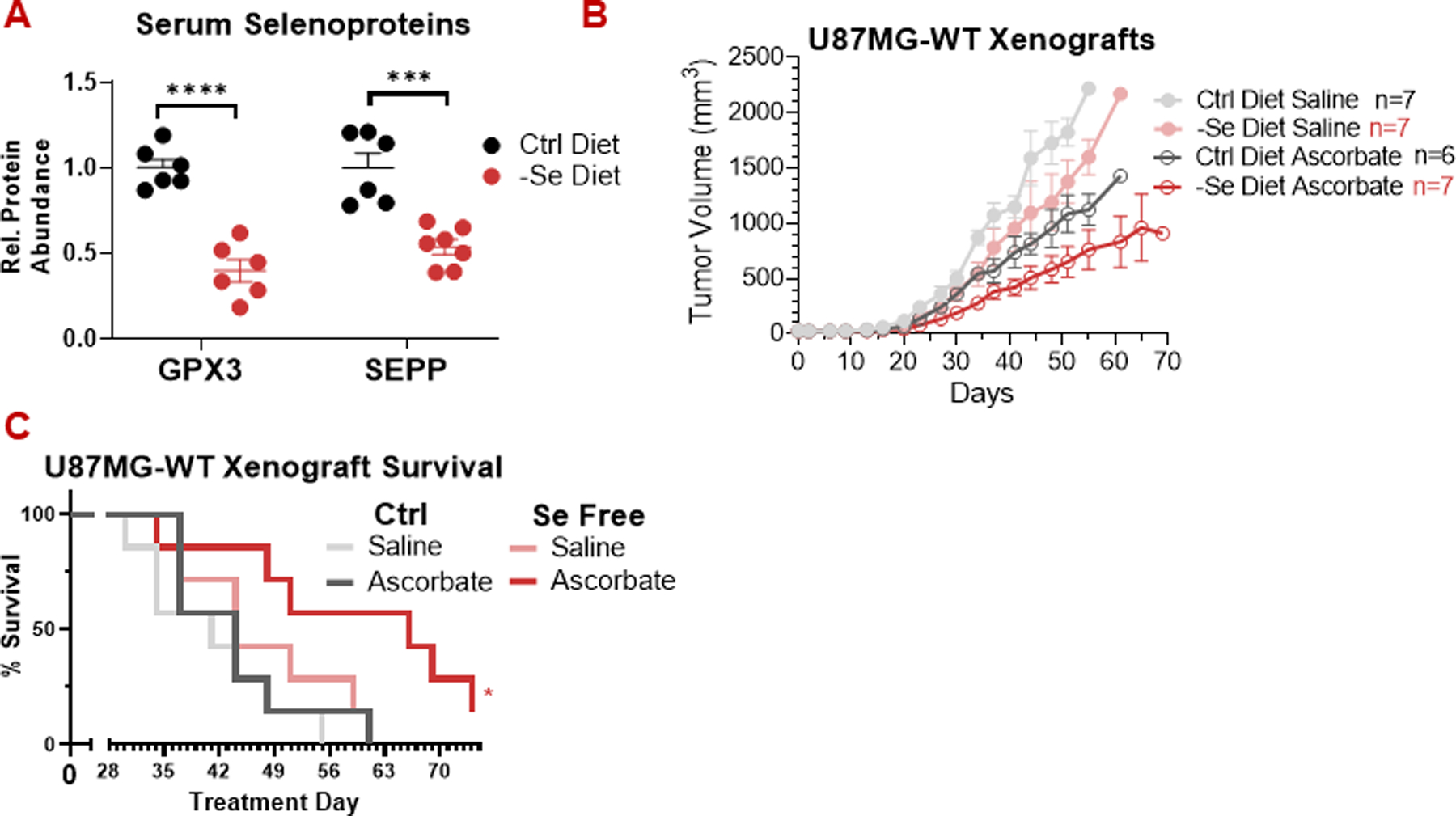
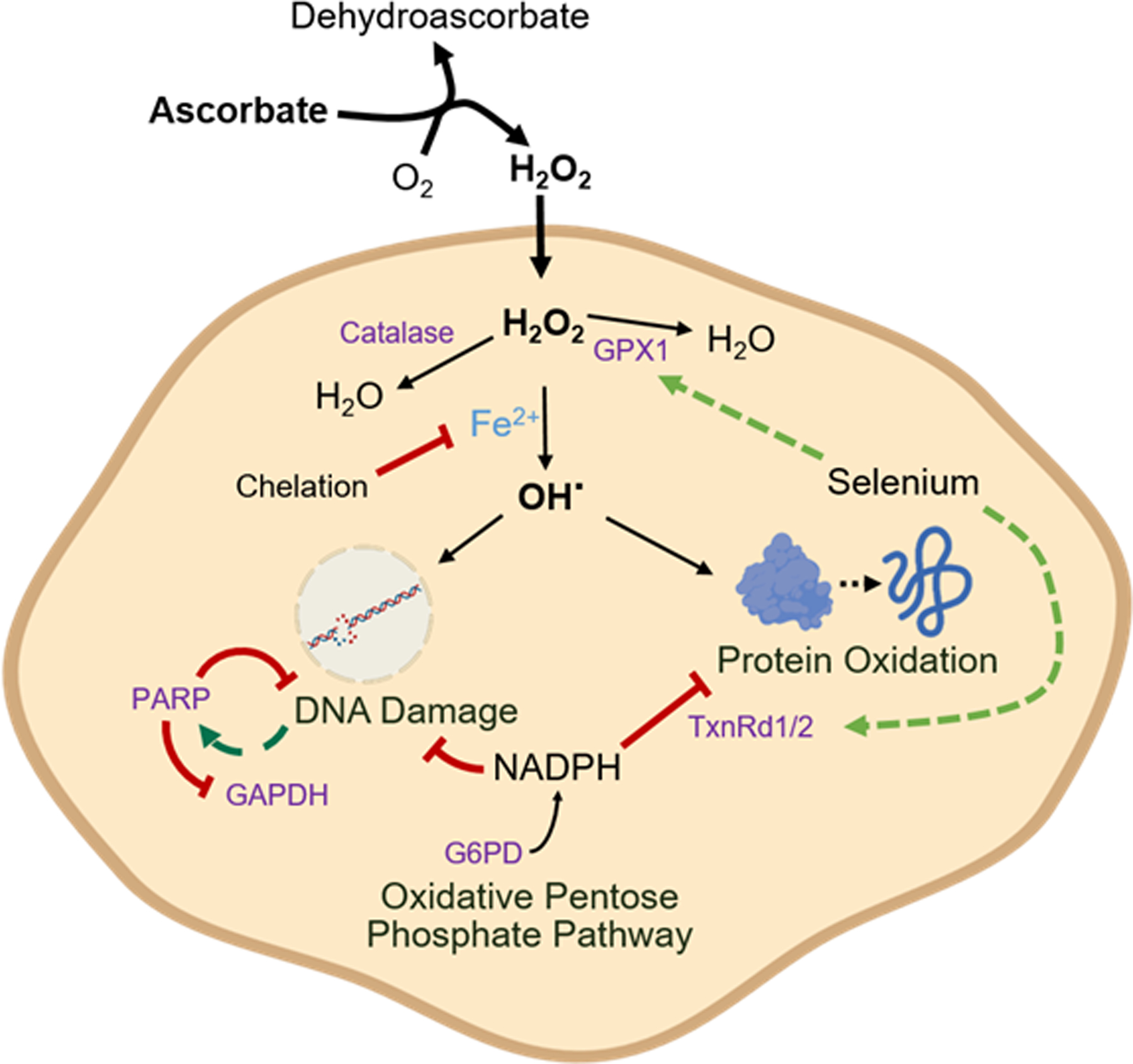
High-dose ascorbate (vitamin C) has shown promising anticancer activity. Two redox mechanisms have been proposed: hydrogen peroxide generation by ascorbate itself or glutathione depletion by dehydroascorbate (formed by ascorbate oxidation). Here we show that the metabolic effects and cytotoxicity of high-dose ascorbate in vitro result from hydrogen peroxide independently of dehydroascorbate. These effects were suppressed by selenium through antioxidant selenoenzymes including glutathione peroxidase 1 (GPX1) but not the classic ferroptosis-inhibiting selenoenzyme GPX4. Selenium-mediated protection from ascorbate was powered by NADPH from the pentose phosphate pathway. In vivo, dietary selenium deficiency resulted in significant enhancement of ascorbate activity against glioblastoma xenografts. These data establish selenoproteins as key mediators of cancer redox homeostasis. Cancer sensitivity to free radical-inducing therapies, including ascorbate, may depend on selenium, providing a dietary approach for improving their anticancer efficacy.
Significance: Selenium restriction augments ascorbate efficacy and extends lifespan in a mouse xenograft model of glioblastoma, suggesting that targeting selenium-mediated antioxidant defenses merits clinical evaluation in combination with ascorbate and other pro-oxidant therapies.
©2022 American Association for Cancer Research.
Conflict of interest statement
Competing Interests
J.D.R. is a paid adviser and/or stockholder in Colorado Research Partners, Kadmon Pharmaceuticals, L.E.A.F. Pharmaceuticals, Rafael Pharmaceuticals and its subsidiaries, Empress, and Agios Pharmaceuticals; a paid consultant of Pfizer; a founder, director, and stockholder of Farber Partners and Serien Therpeutics. C.S.R.J. declares no conflicts of interest.
Figures
Similar articles
-
Selenium supplementation protects cancer cells from the oxidative stress and cytotoxicity induced by the combination of ascorbate and menadione sodium bisulfite.Free Radic Biol Med. 2025 Jun;233:317-329. doi: 10.1016/j.freeradbiomed.2025.03.049. Epub 2025 Apr 1. Free Radic Biol Med. 2025. PMID: 40180024
-
Interference of selenium and selenoproteins with the insulin-regulated carbohydrate and lipid metabolism.Free Radic Biol Med. 2013 Dec;65:1538-1547. doi: 10.1016/j.freeradbiomed.2013.07.016. Epub 2013 Jul 18. Free Radic Biol Med. 2013. PMID: 23872396 Review.
-
Selective up-regulation of human selenoproteins in response to oxidative stress.J Biol Chem. 2014 May 23;289(21):14750-61. doi: 10.1074/jbc.M114.551994. Epub 2014 Apr 4. J Biol Chem. 2014. PMID: 24706762 Free PMC article.
-
Glutathione peroxidase-1 gene knockout on body antioxidant defense in mice.Biofactors. 2001;14(1-4):93-9. doi: 10.1002/biof.5520140113. Biofactors. 2001. PMID: 11568445 Review.
-
Stimulation of the pentose phosphate pathway and glutathione levels by dehydroascorbate, the oxidized form of vitamin C.FASEB J. 2000 Jul;14(10):1352-61. doi: 10.1096/fj.14.10.1352. FASEB J. 2000. PMID: 10877828
Cited by
-
Mechanisms of Vitamins Inhibiting Ferroptosis.Antioxidants (Basel). 2024 Dec 20;13(12):1571. doi: 10.3390/antiox13121571. Antioxidants (Basel). 2024. PMID: 39765898 Free PMC article. Review.
-
A new perspective on iron-dependent cell death: PRDX-1-mediated ferroptosis in tumor cells.Apoptosis. 2025 Aug;30(7-8):1645-1664. doi: 10.1007/s10495-025-02129-6. Epub 2025 Jun 9. Apoptosis. 2025. PMID: 40488836 Review.
-
A Diet Lacking Selenium, but Not Zinc, Copper or Manganese, Induces Anticancer Activity in Mice with Metastatic Cancers.Nutrients. 2024 Jul 12;16(14):2249. doi: 10.3390/nu16142249. Nutrients. 2024. PMID: 39064692 Free PMC article.
-
Dehydroascorbic acid sensitizes cancer cells to system xc- inhibition-induced ferroptosis by promoting lipid droplet peroxidation.Cell Death Dis. 2023 Sep 27;14(9):637. doi: 10.1038/s41419-023-06153-9. Cell Death Dis. 2023. PMID: 37752118 Free PMC article.
-
Organic Selenium induces ferroptosis in pancreatic cancer cells.Redox Biol. 2023 Dec;68:102962. doi: 10.1016/j.redox.2023.102962. Epub 2023 Nov 20. Redox Biol. 2023. PMID: 38029455 Free PMC article.
References
-
- Ching SYL, Prins AW, and Beilby JP. Stability of ascorbic acid in serum and plasma prior to analysis. Ann Clin Biochem 2002; 39: 518–20 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
