

A cross-disorder dosage sensitivity map of the human genome
- PMID: 35917817
- PMCID: PMC9742861
- DOI: 10.1016/j.cell.2022.06.036
A cross-disorder dosage sensitivity map of the human genome
Abstract
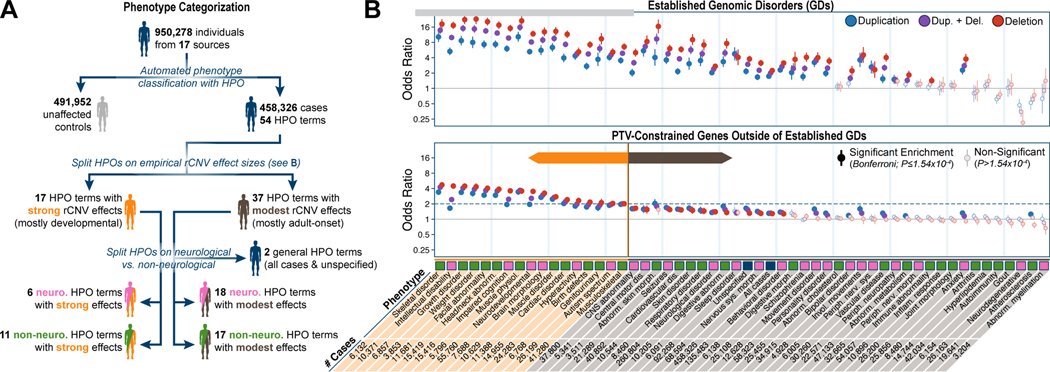
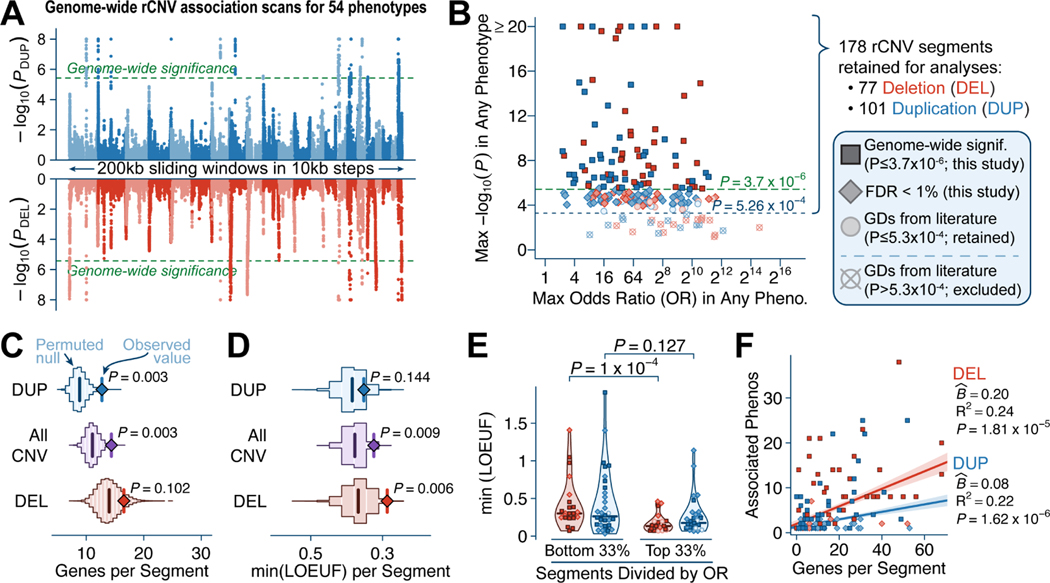
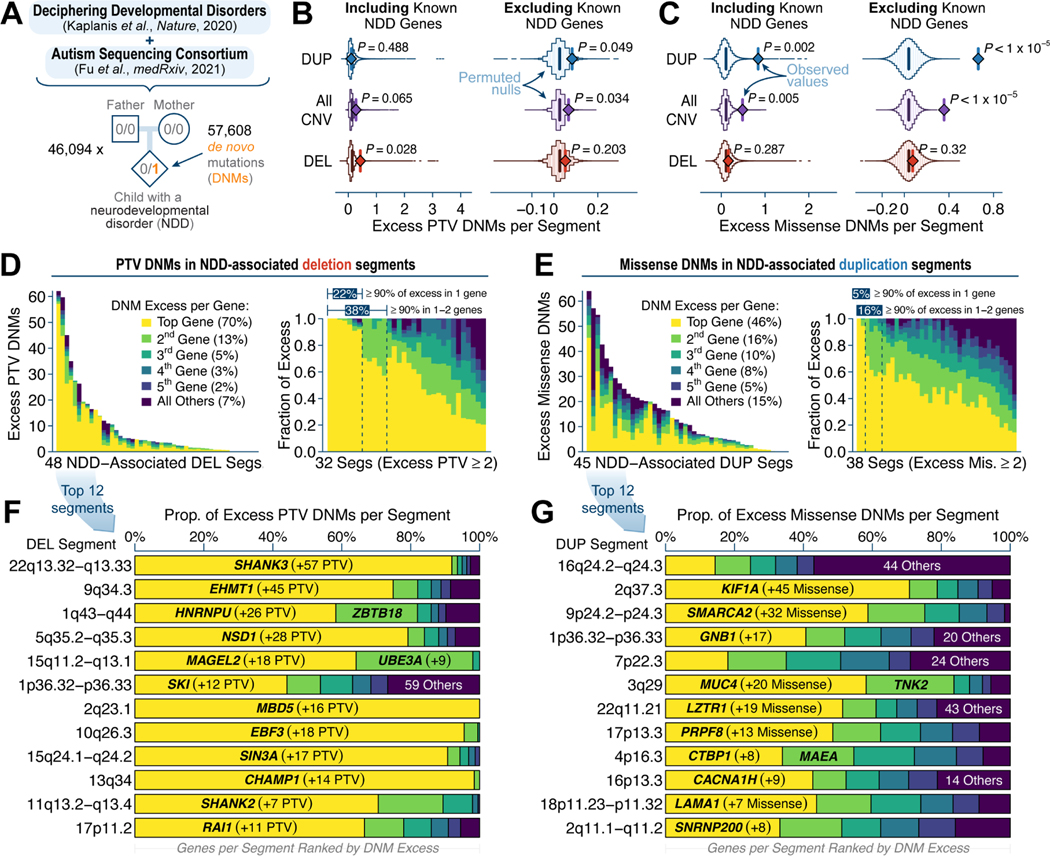
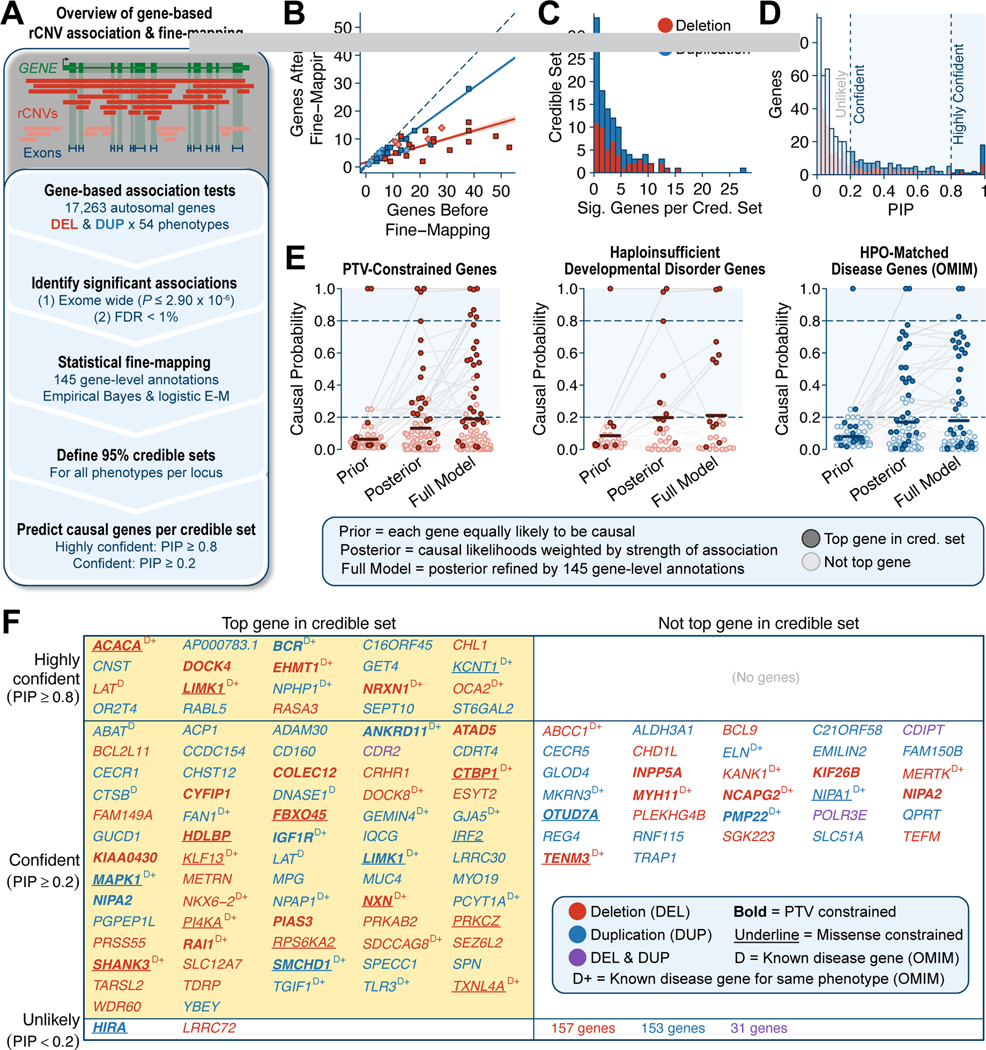
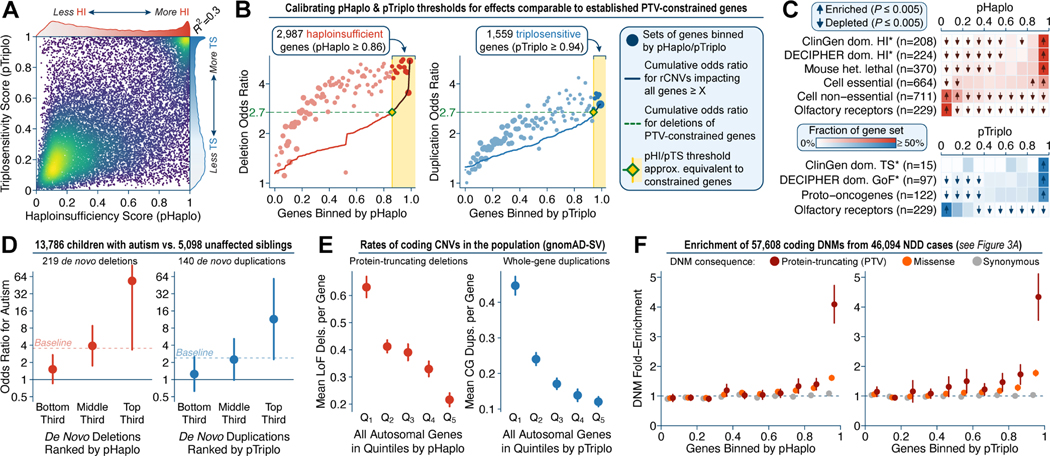
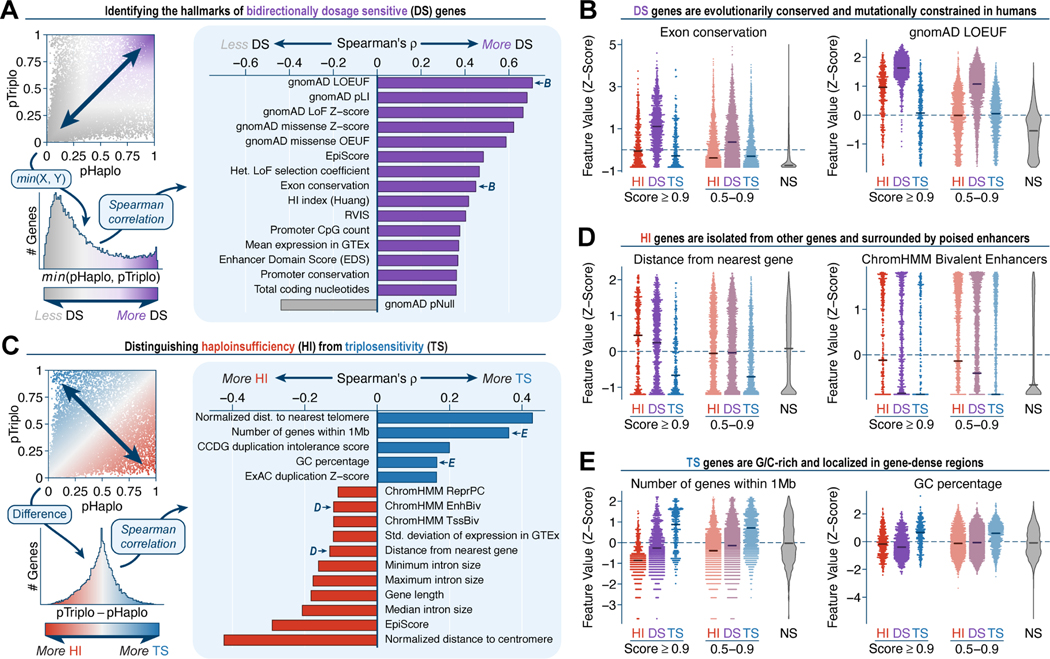
Rare copy-number variants (rCNVs) include deletions and duplications that occur infrequently in the global human population and can confer substantial risk for disease. In this study, we aimed to quantify the properties of haploinsufficiency (i.e., deletion intolerance) and triplosensitivity (i.e., duplication intolerance) throughout the human genome. We harmonized and meta-analyzed rCNVs from nearly one million individuals to construct a genome-wide catalog of dosage sensitivity across 54 disorders, which defined 163 dosage sensitive segments associated with at least one disorder. These segments were typically gene dense and often harbored dominant dosage sensitive driver genes, which we were able to prioritize using statistical fine-mapping. Finally, we designed an ensemble machine-learning model to predict probabilities of dosage sensitivity (pHaplo & pTriplo) for all autosomal genes, which identified 2,987 haploinsufficient and 1,559 triplosensitive genes, including 648 that were uniquely triplosensitive. This dosage sensitivity resource will provide broad utility for human disease research and clinical genetics.
Keywords: copy-number variation; developmental disorders; disease association; dosage sensitivity; genomics; haploinsufficiency; statistical genetics; structural variation; triplosensitivity.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.E.T. receives research funding and/or reagents from Levo Therapeutics, Microsoft Inc., and Illumina Inc. R.B., C. Lauricella, A.J., L.M., S.W., and J.M. are employees of GeneDx, Inc. S.A. is an employee of Invitae Corp.
Figures

Comment in
-
The gene dose makes the disease.Cell. 2022 Aug 4;185(16):2850-2852. doi: 10.1016/j.cell.2022.07.005. Cell. 2022. PMID: 35931018 Free PMC article.
-
Mapping dosage.Nat Rev Genet. 2022 Oct;23(10):583. doi: 10.1038/s41576-022-00528-y. Nat Rev Genet. 2022. PMID: 36042286 No abstract available.
References
-
- Abuzzahab MJ., Schneider A., Goddard A., Grigorescu F., Lautier C., Keller E., Kiess W., Klammt J., Kratzsch J., Osgood D., et al.. (2003). IGF-I receptor mutations resulting in intrauterine and postnatal growth retardation. The New England journal of medicine 349, 2211–2222. - PubMed
-
- Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, et al. (2012). Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44, 435–439, s431–432. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
- R03 HD099547/HD/NICHD NIH HHS/United States
- L30 NS093200/NS/NINDS NIH HHS/United States
- T32 HG002295/HG/NHGRI NIH HHS/United States
- K08 NS117891/NS/NINDS NIH HHS/United States
- R01 HD081256/HD/NICHD NIH HHS/United States
- R01 DE031261/DE/NIDCR NIH HHS/United States
- R56 MH115957/MH/NIMH NIH HHS/United States
- R01 NS093200/NS/NINDS NIH HHS/United States
- P50 HD104224/HD/NICHD NIH HHS/United States
- R01 MH115957/MH/NIMH NIH HHS/United States
- R00 DE026824/DE/NIDCR NIH HHS/United States
- R01 NS102423/NS/NINDS NIH HHS/United States
- R01 MH106826/MH/NIMH NIH HHS/United States
- R01 HD096326/HD/NICHD NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
