

A SOX2-engineered epigenetic silencer factor represses the glioblastoma genetic program and restrains tumor development
- PMID: 35921410
- PMCID: PMC9348799
- DOI: 10.1126/sciadv.abn3986
A SOX2-engineered epigenetic silencer factor represses the glioblastoma genetic program and restrains tumor development
Abstract
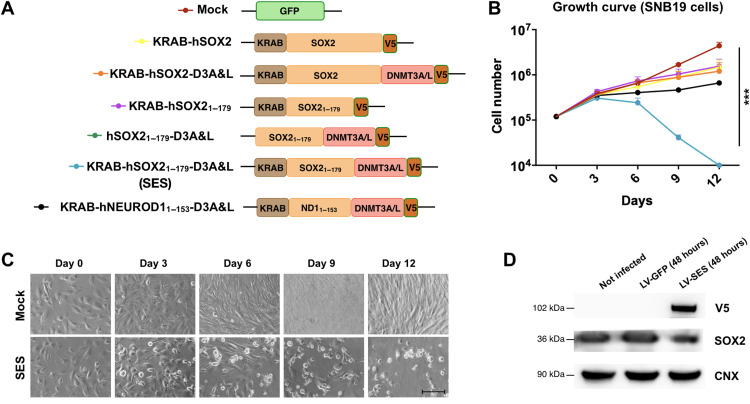
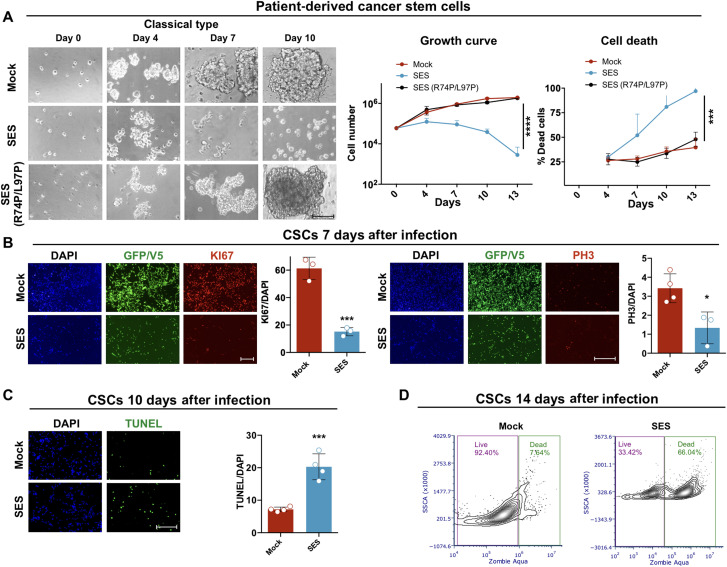
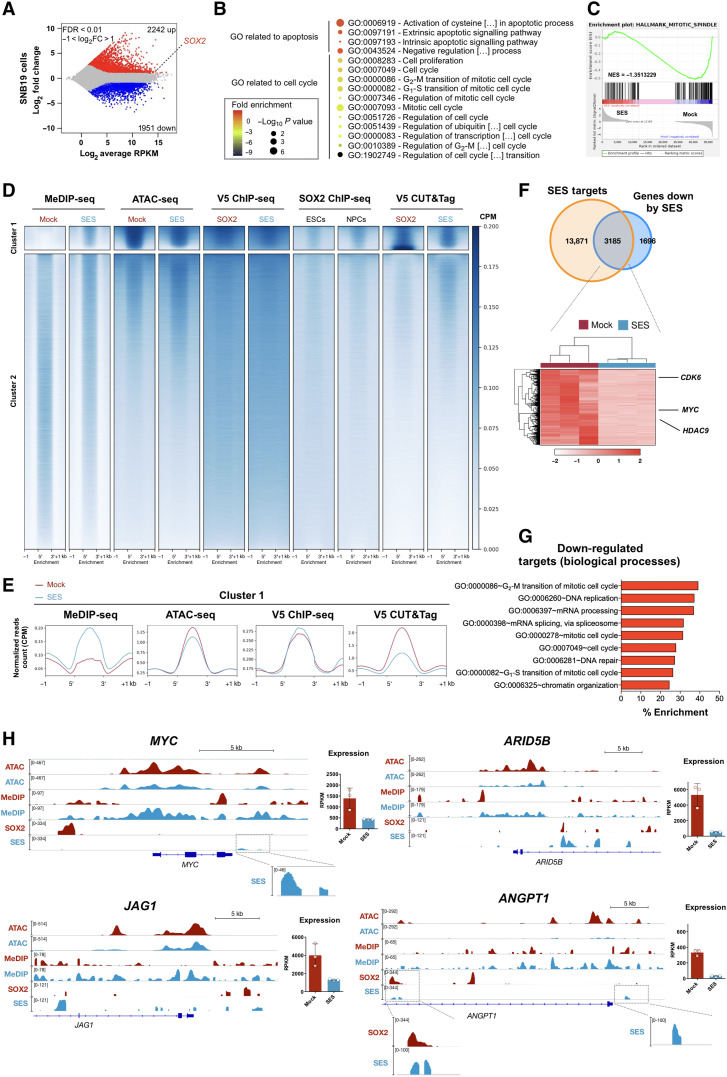
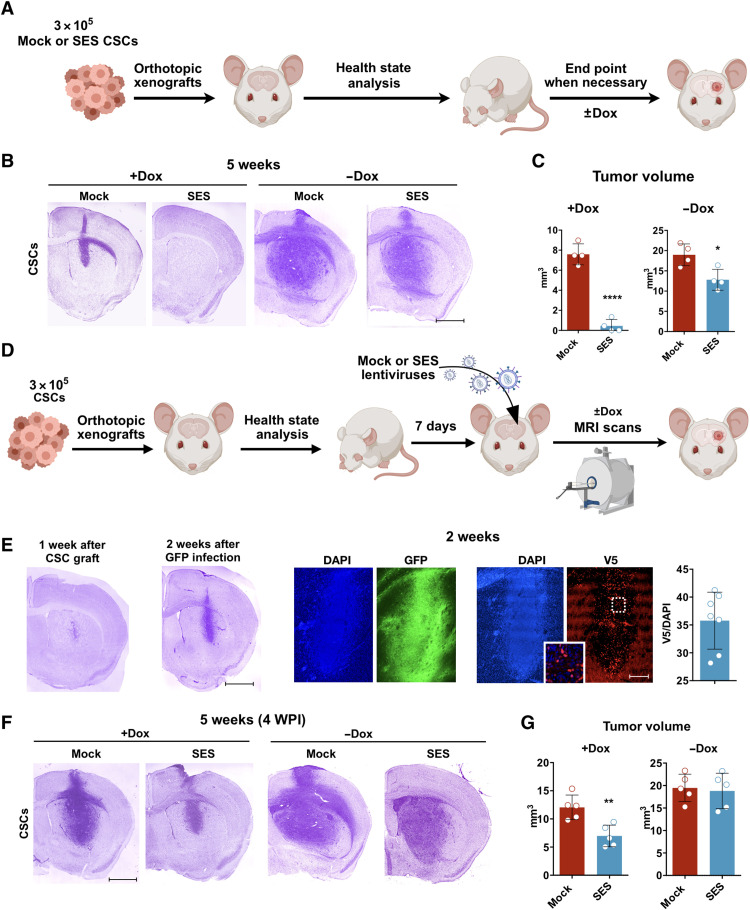
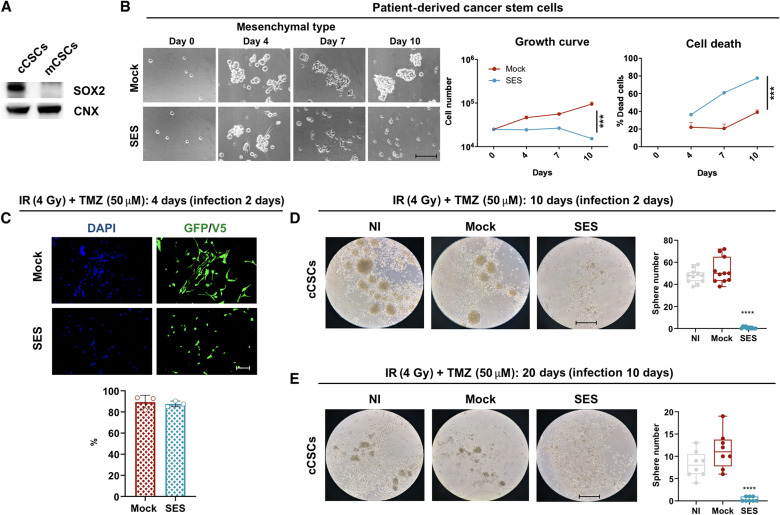
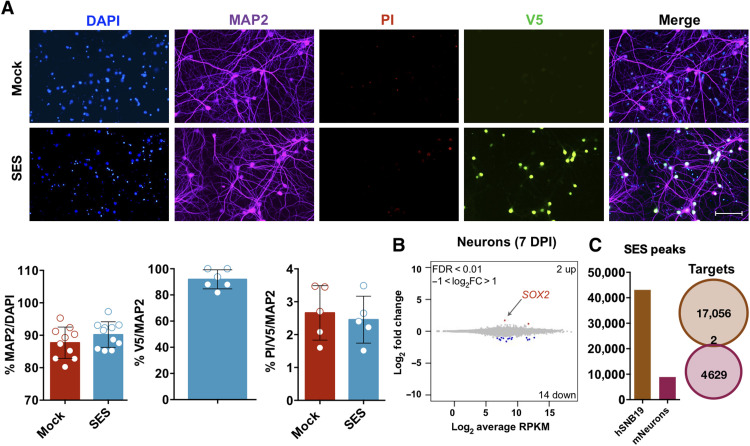
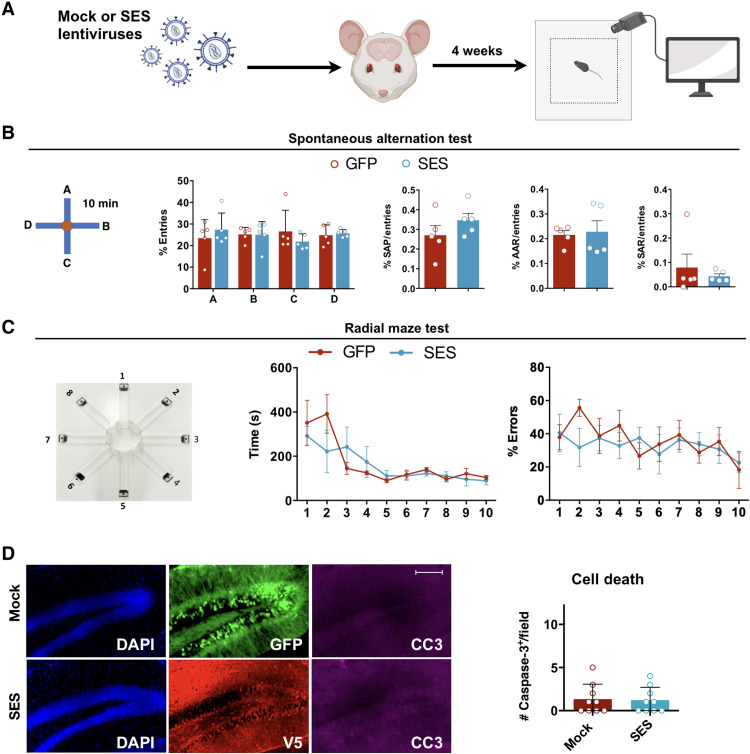
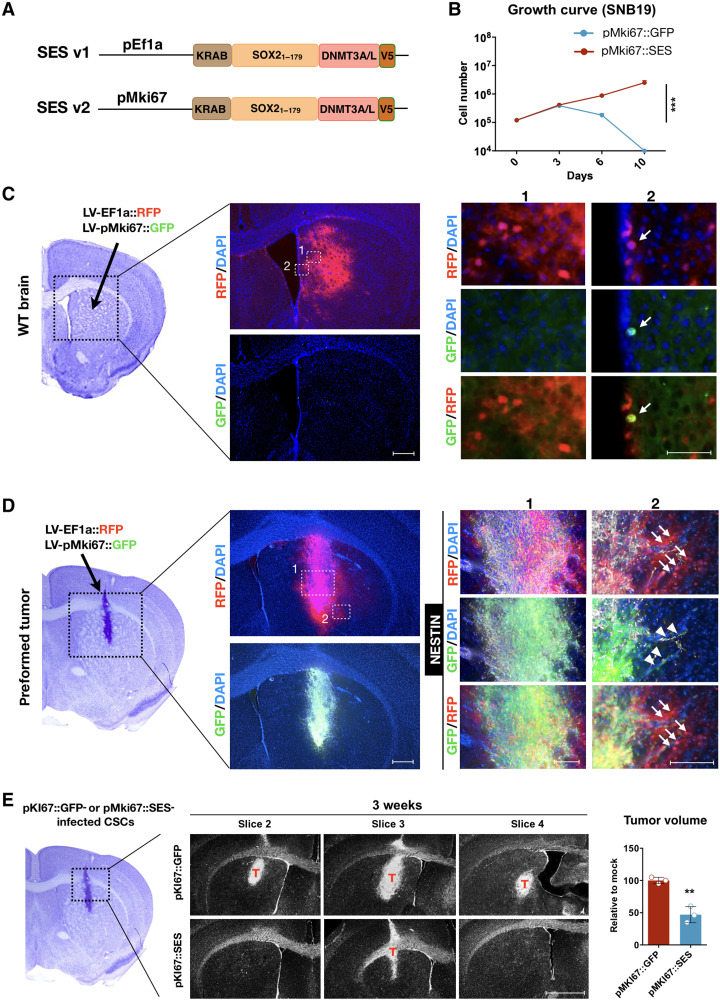
Current therapies remain unsatisfactory in preventing the recurrence of glioblastoma multiforme (GBM), which leads to poor patient survival. By rational engineering of the transcription factor SOX2, a key promoter of GBM malignancy, together with the Kruppel-associated box and DNA methyltransferase3A/L catalytic domains, we generated a synthetic repressor named SOX2 epigenetic silencer (SES), which induces the transcriptional silencing of its original targets. By doing so, SES kills both glioma cell lines and patient-derived cancer stem cells in vitro and in vivo. SES expression, through local viral delivery in mouse xenografts, induces strong regression of human tumors and survival rescue. Conversely, SES is not harmful to neurons and glia, also thanks to a minimal promoter that restricts its expression in mitotically active cells, rarely present in the brain parenchyma. Collectively, SES produces a significant silencing of a large fraction of the SOX2 transcriptional network, achieving high levels of efficacy in repressing aggressive brain tumors.
Figures
References
-
- Stupp R., Mason W. P., van den Bent M. J., Weller M., Fisher B., Taphoorn M. J., Belanger K., Brandes A. A., Marosi C., Bogdahn U., Curschmann J., Janzer R. C., Ludwin S. K., Gorlia T., Allgeier A., Lacombe D., Cairncross J. G., Eisenhauer E., Mirimanoff R. O.; European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group , Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005). - PubMed
-
- Delgado-López P. D., Corrales-García E. M., Survival in glioblastoma: A review on the impact of treatment modalities. Clin. Transl. Oncol. 18, 1062–1071 (2016). - PubMed
-
- Singh S. K., Hawkins C., Clarke I. D., Squire J. A., Bayani J., Hide T., Henkelman R. M., Cusimano M. D., Dirks P. B., Identification of human brain tumour initiating cells. Nature 432, 396–401 (2004). - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
