

Transcriptome variation in human tissues revealed by long-read sequencing
- PMID: 35922509
- PMCID: PMC10337767
- DOI: 10.1038/s41586-022-05035-y
Transcriptome variation in human tissues revealed by long-read sequencing
Abstract
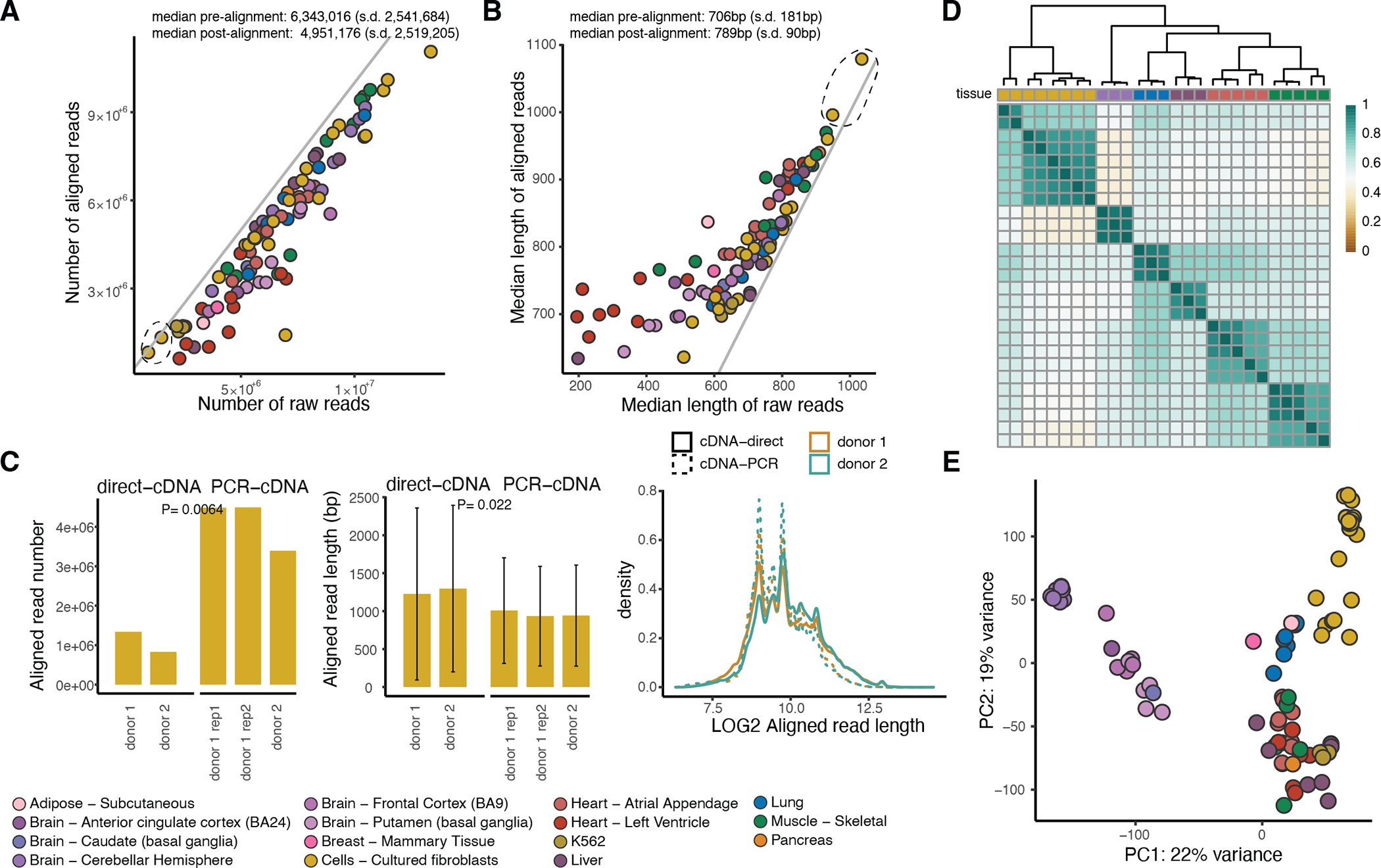
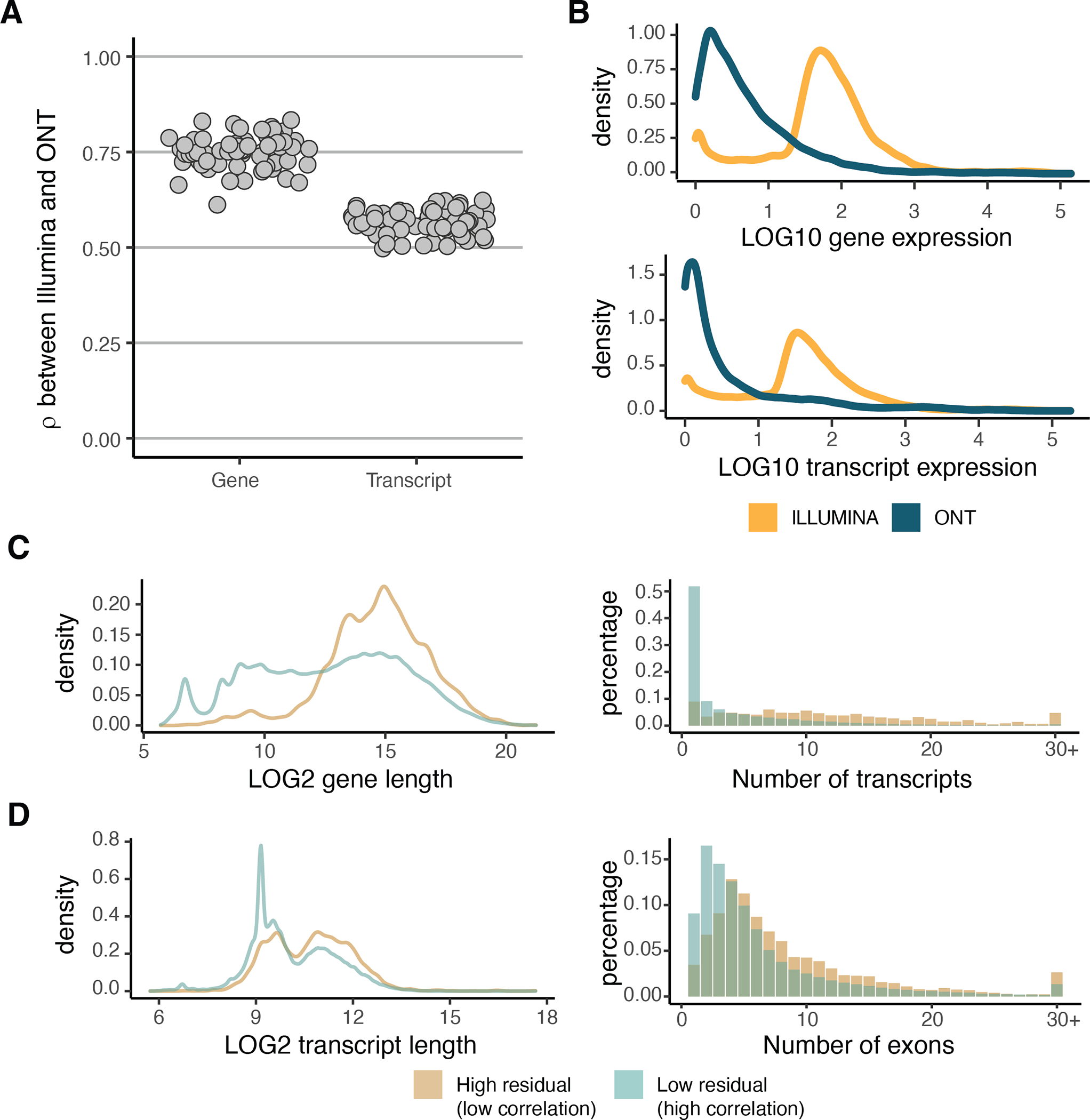
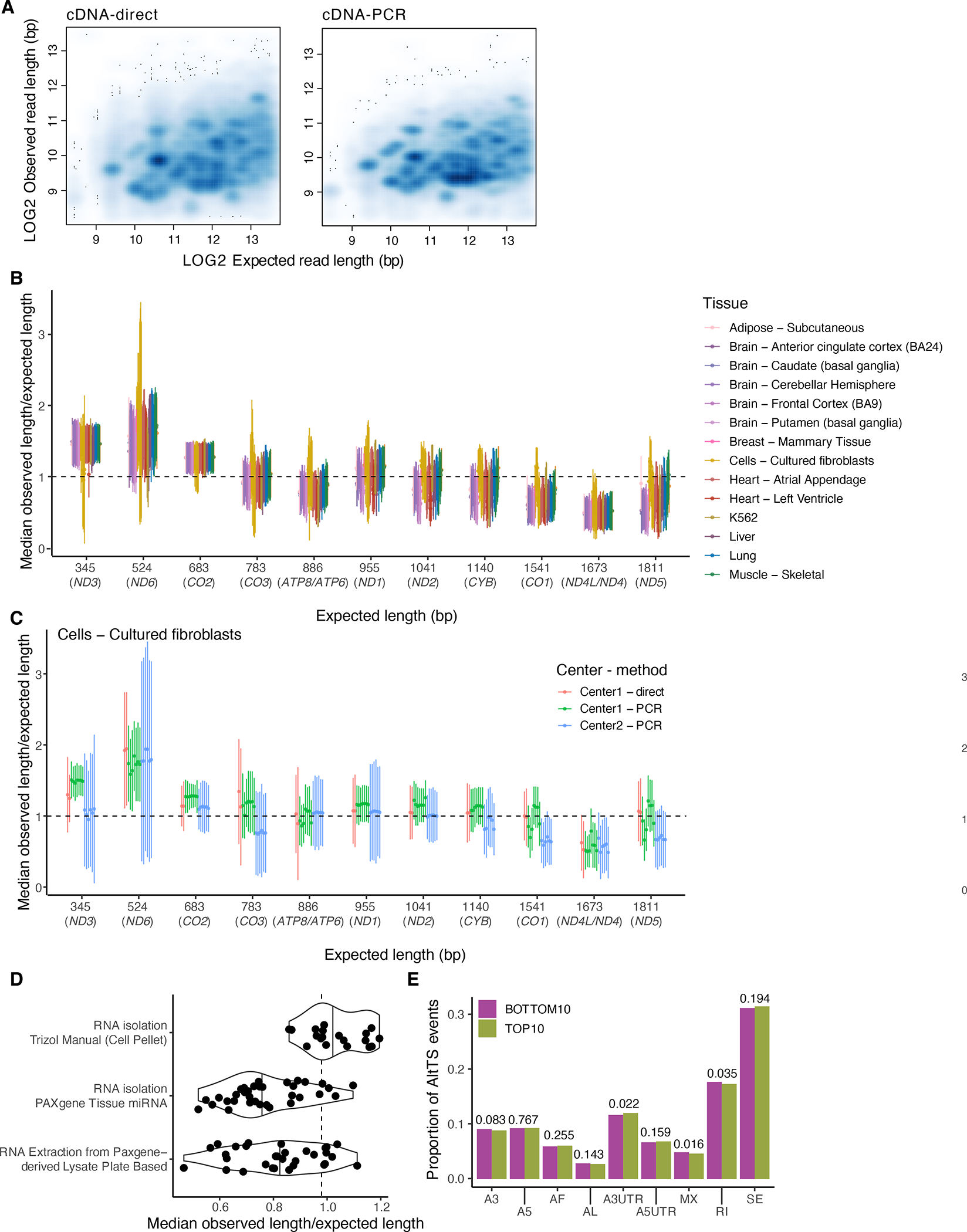
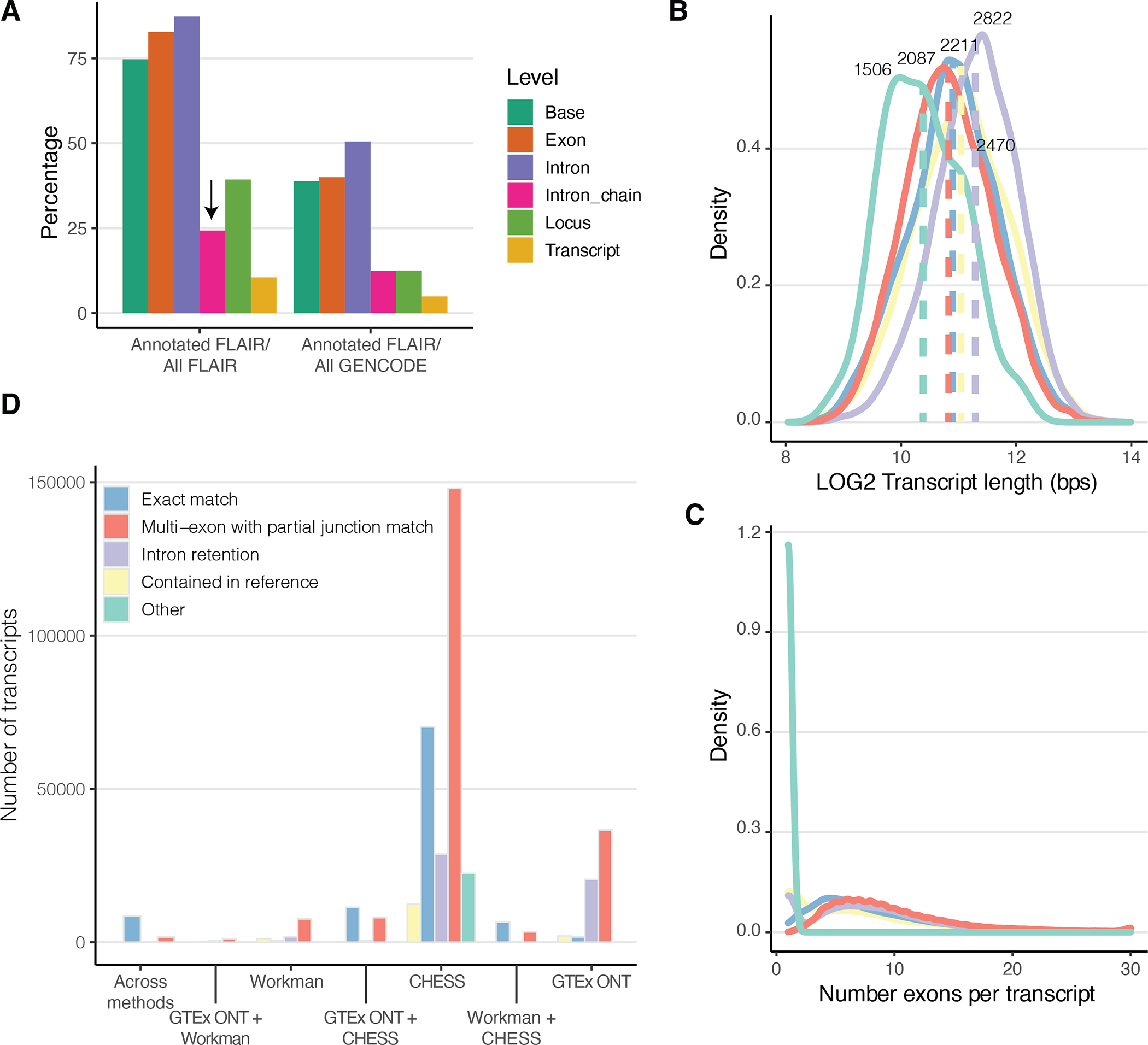
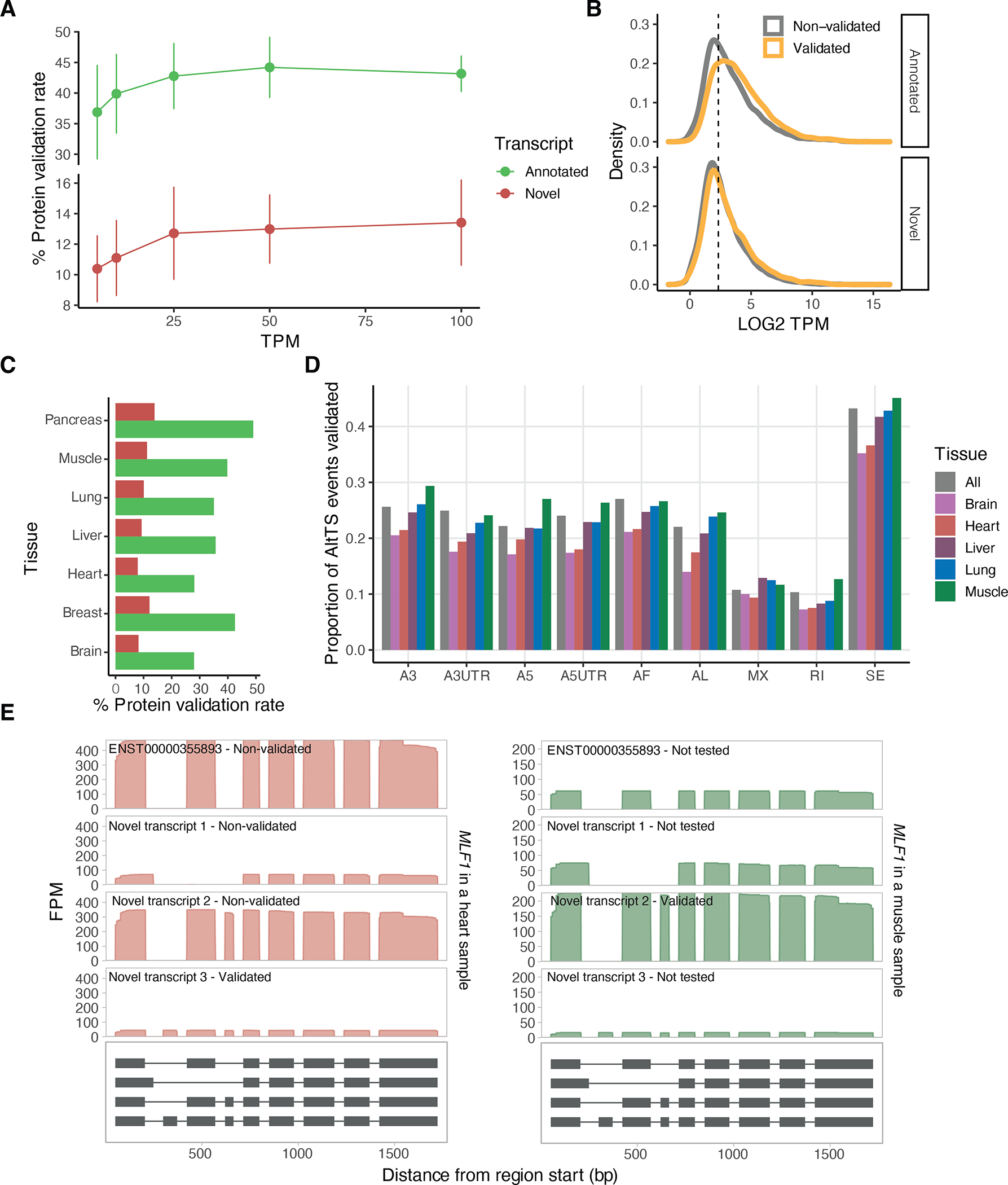
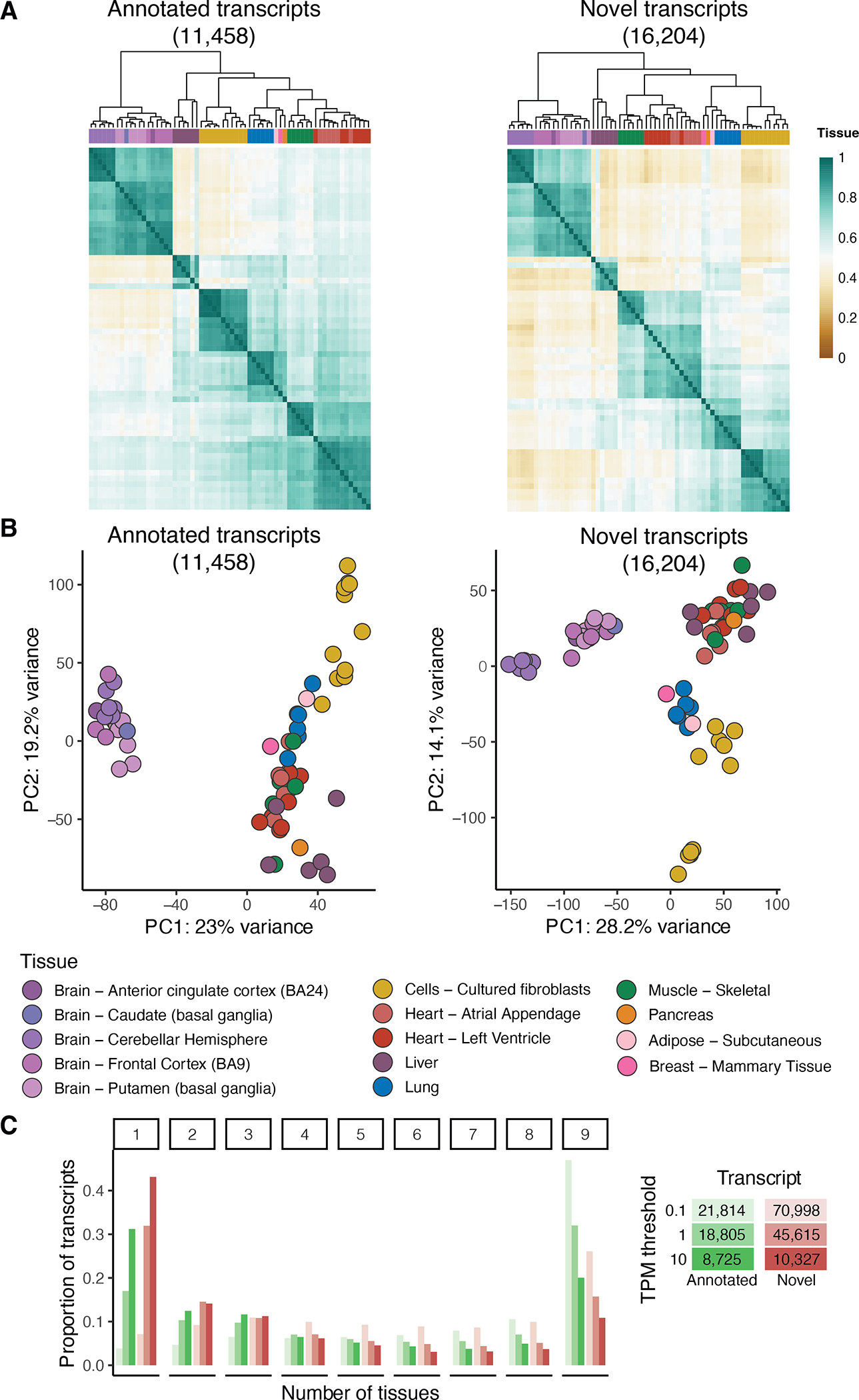
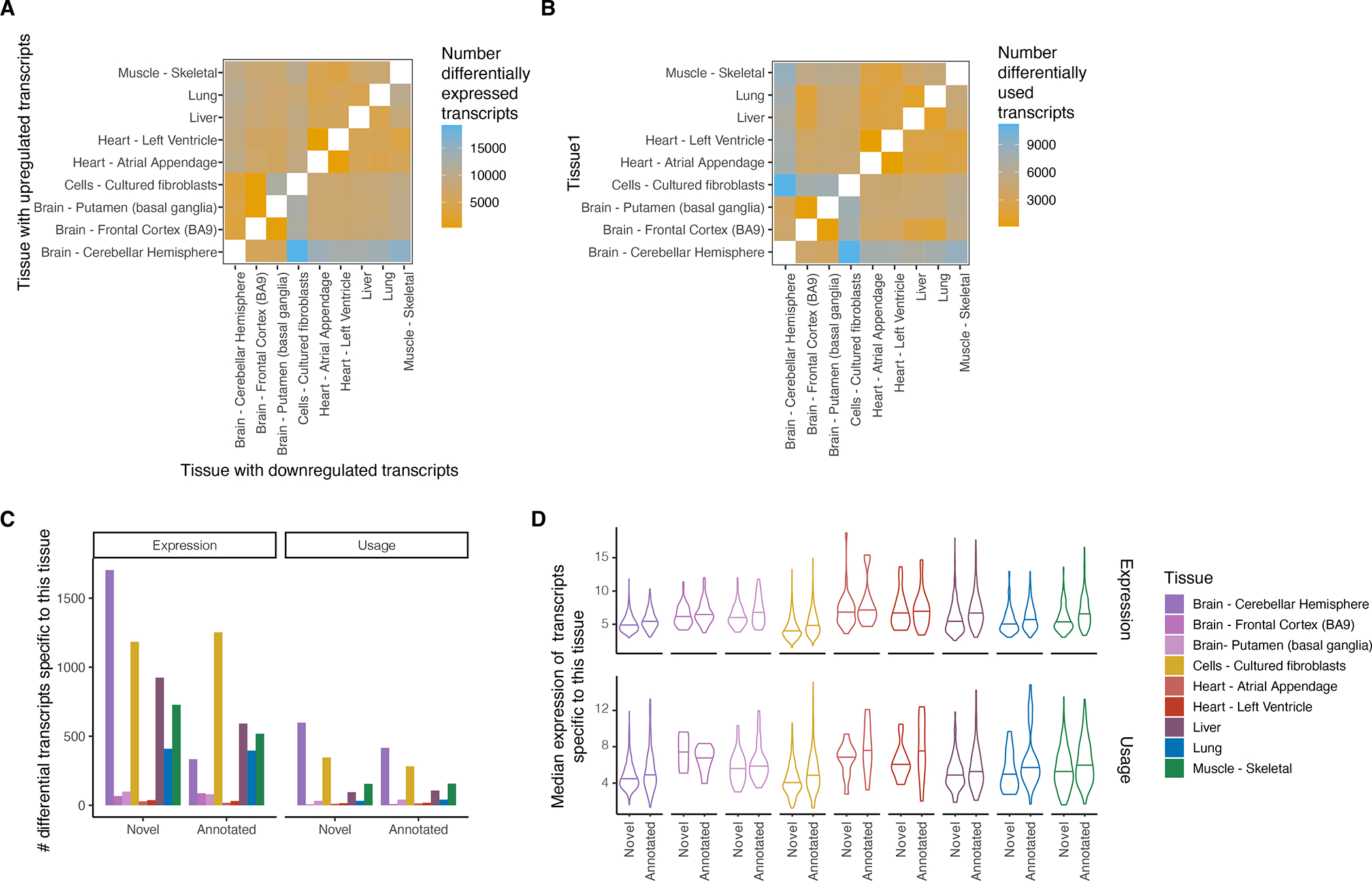
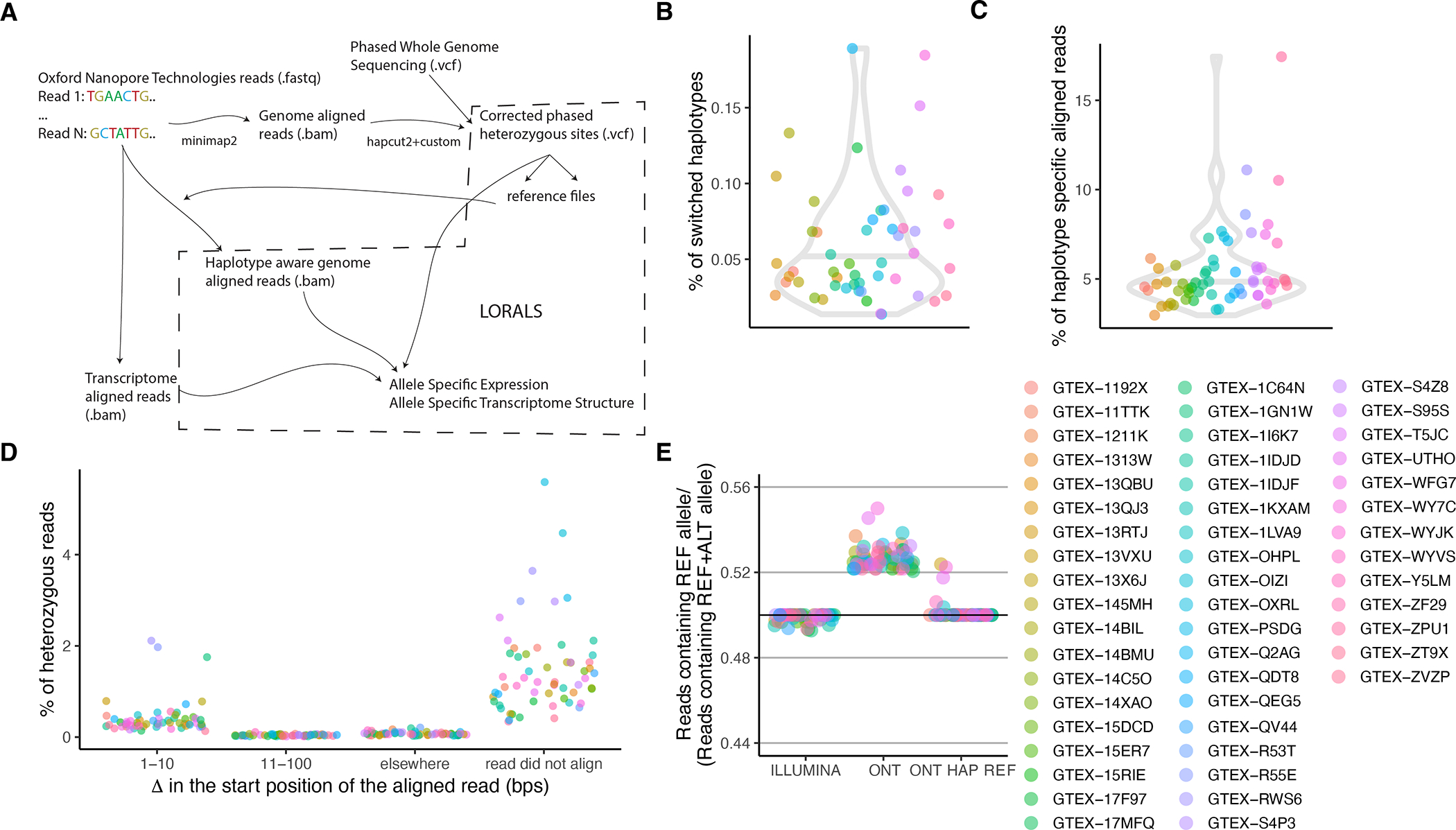
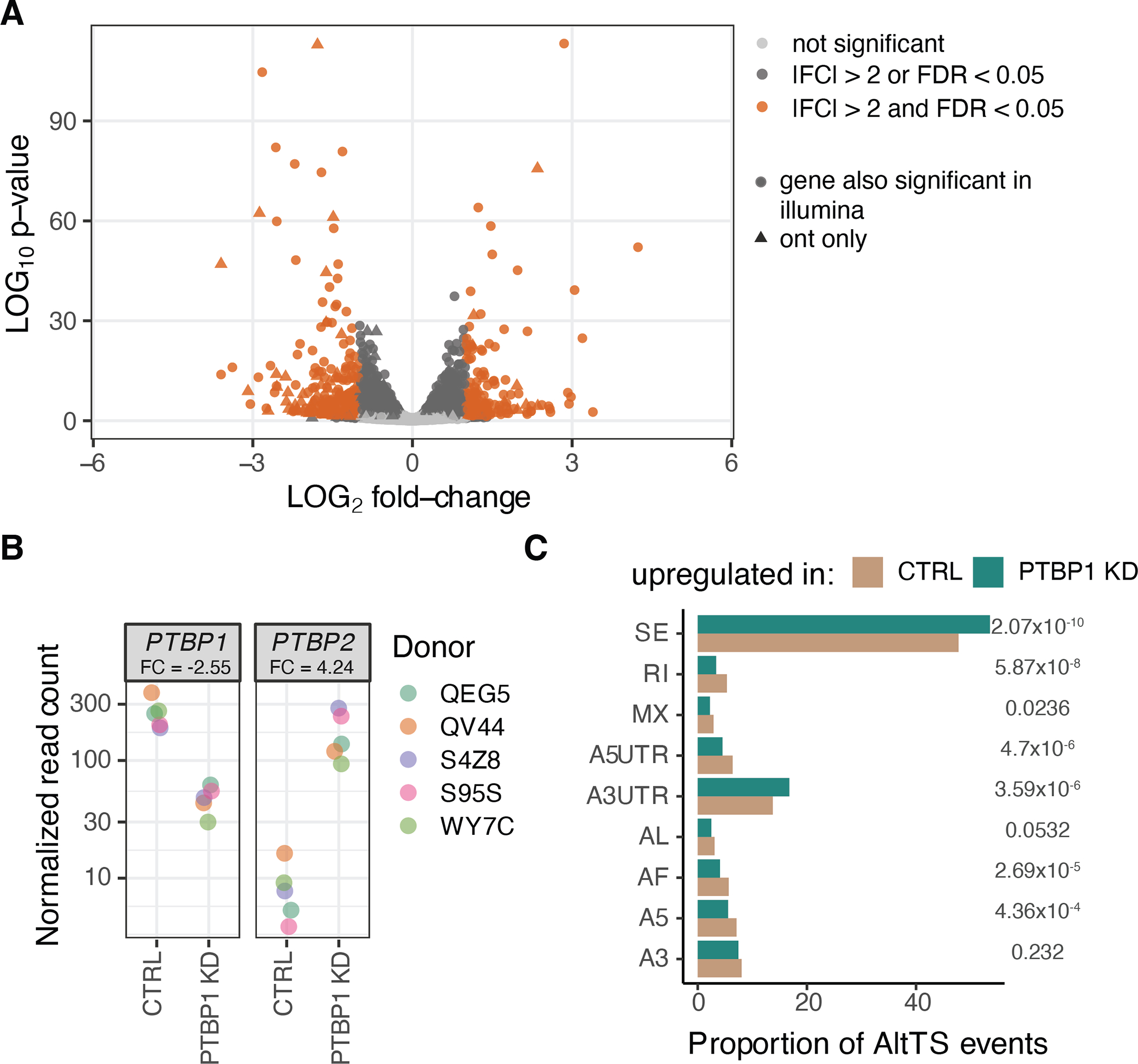
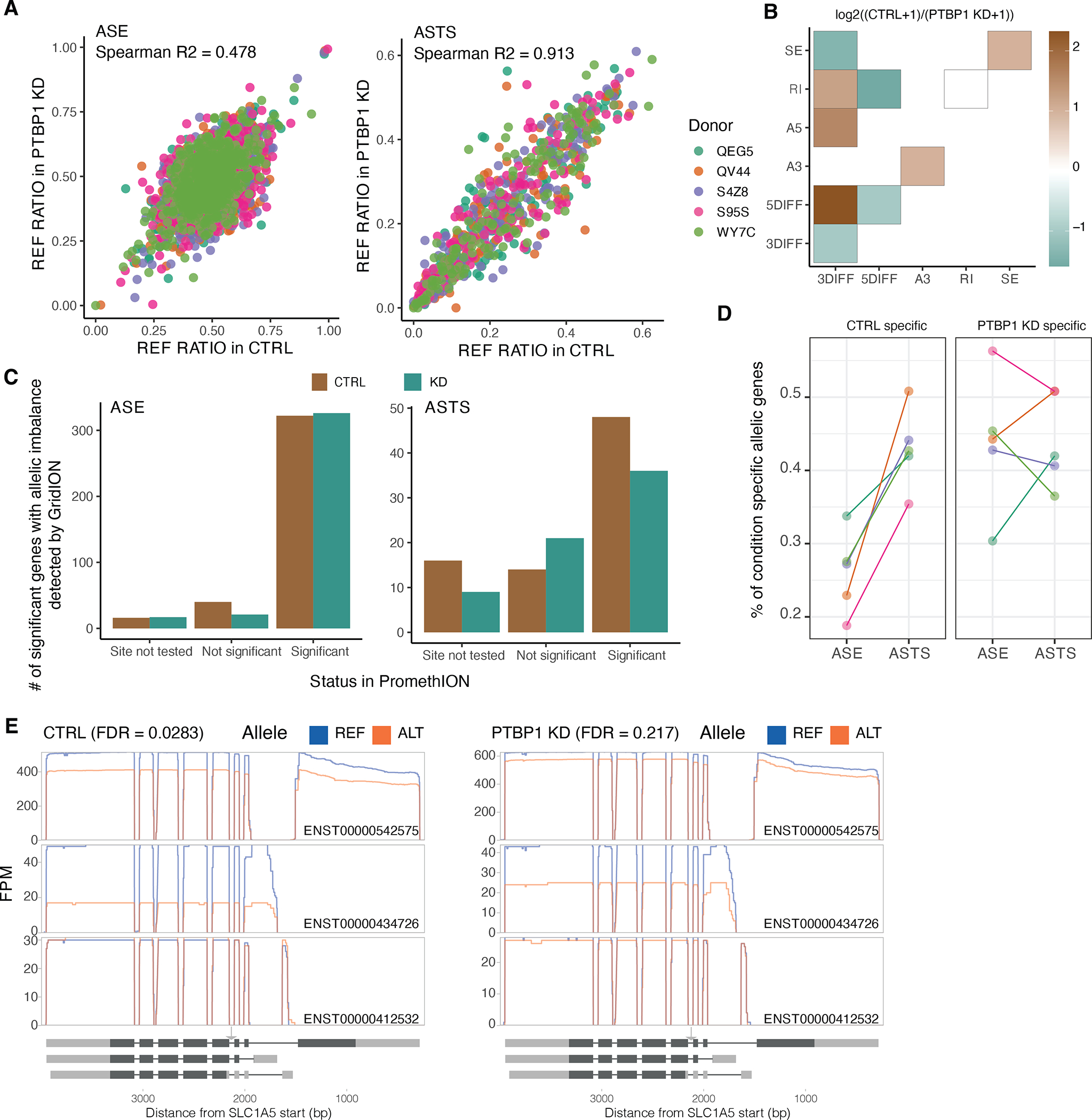
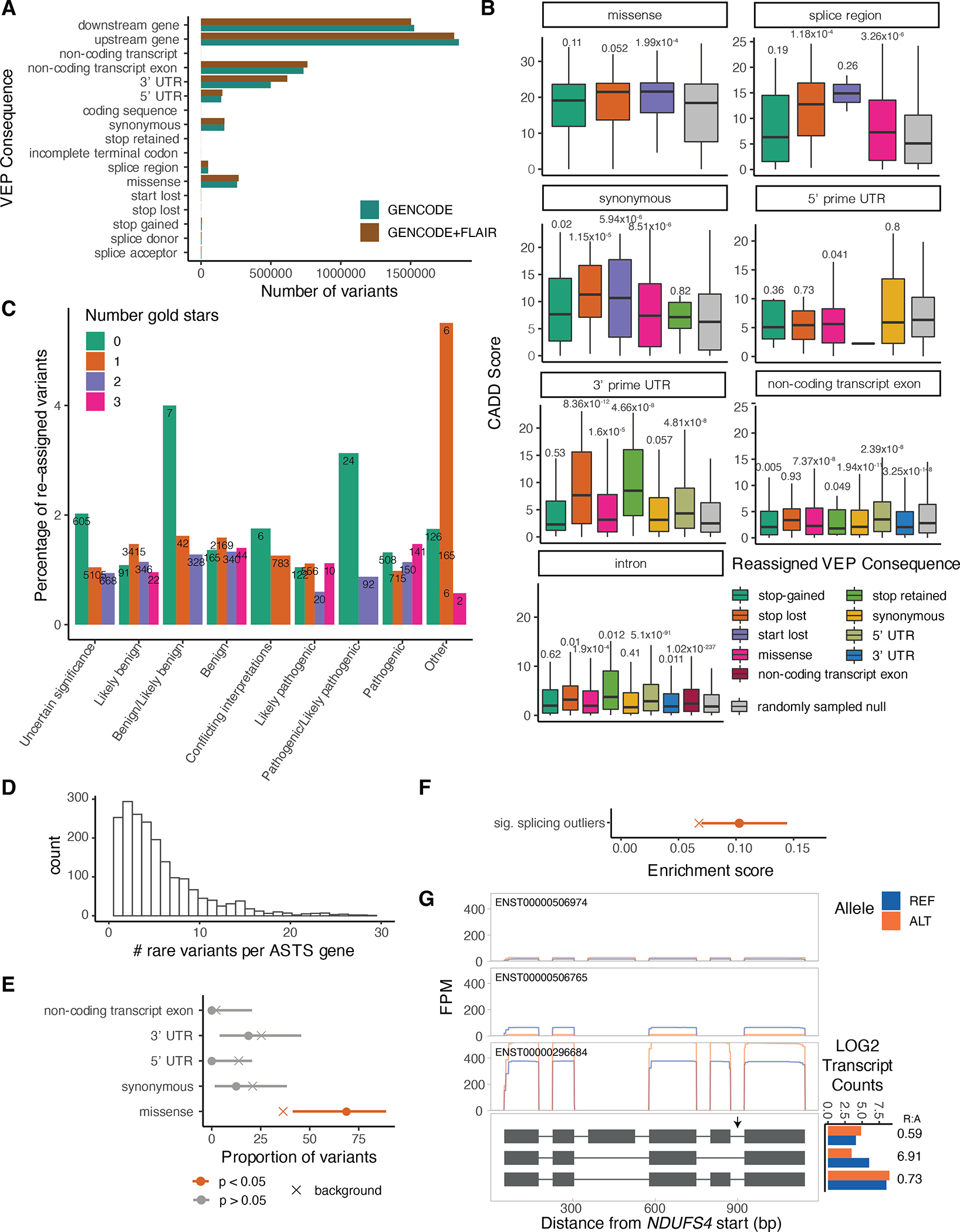
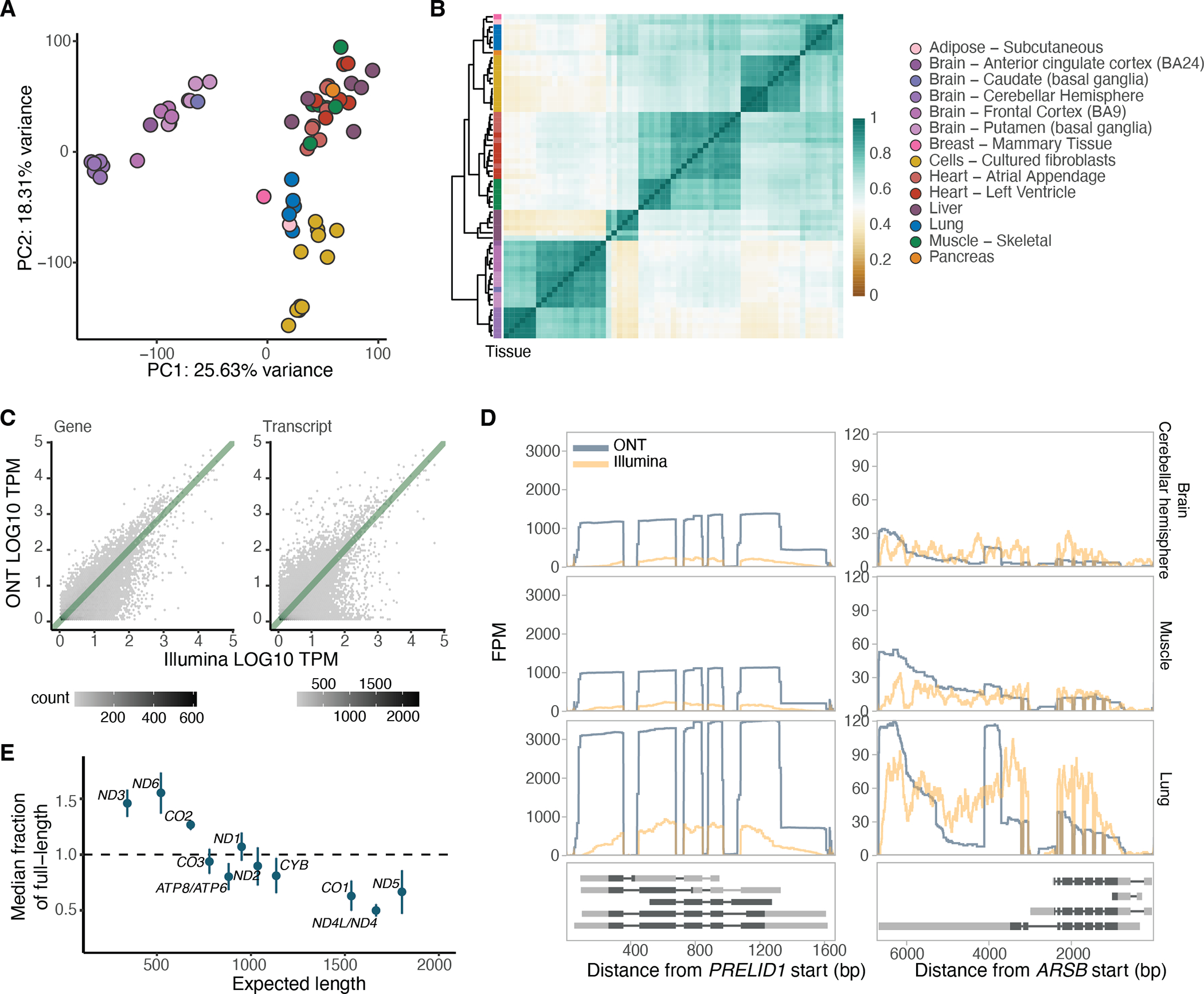
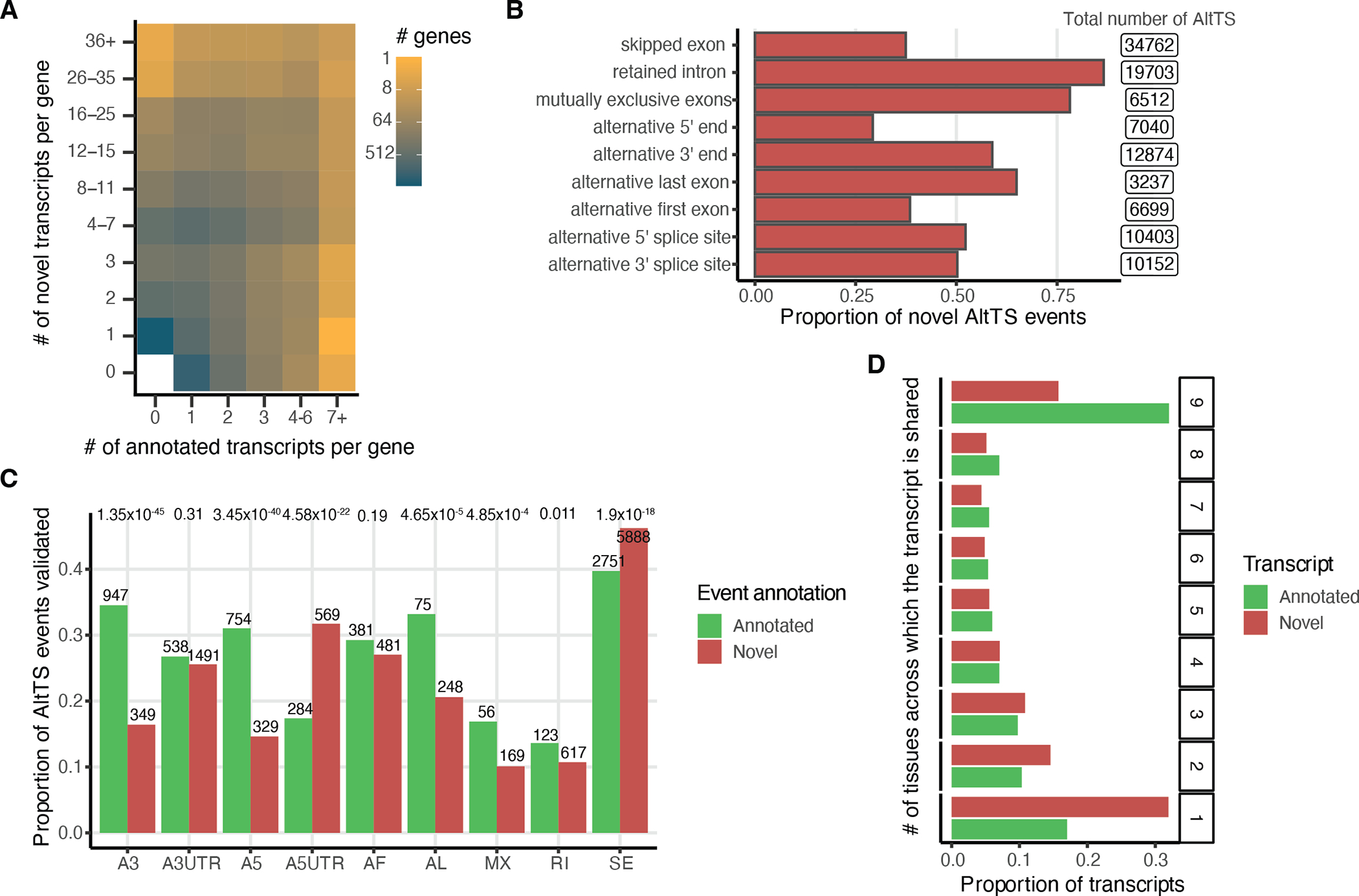
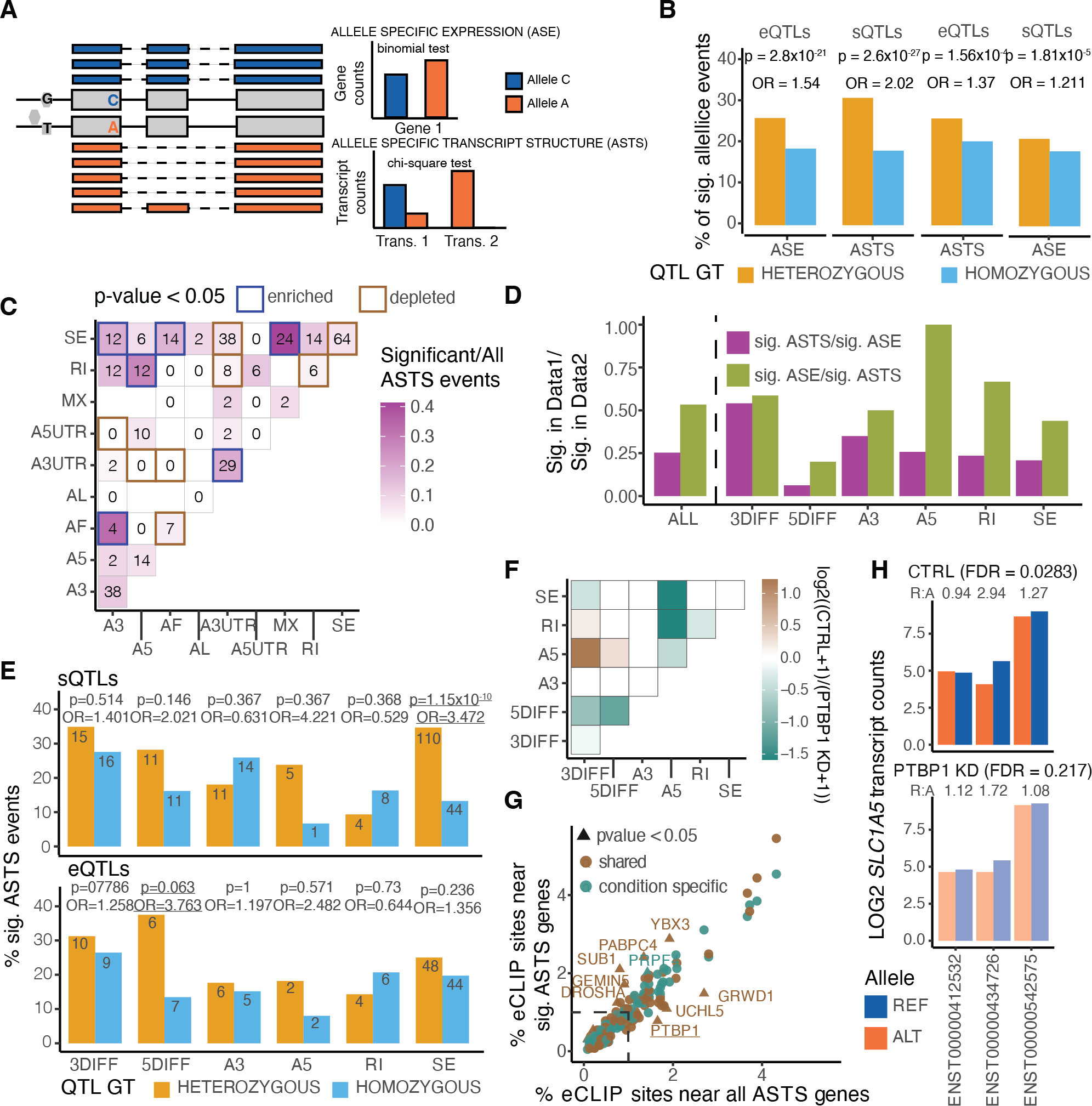
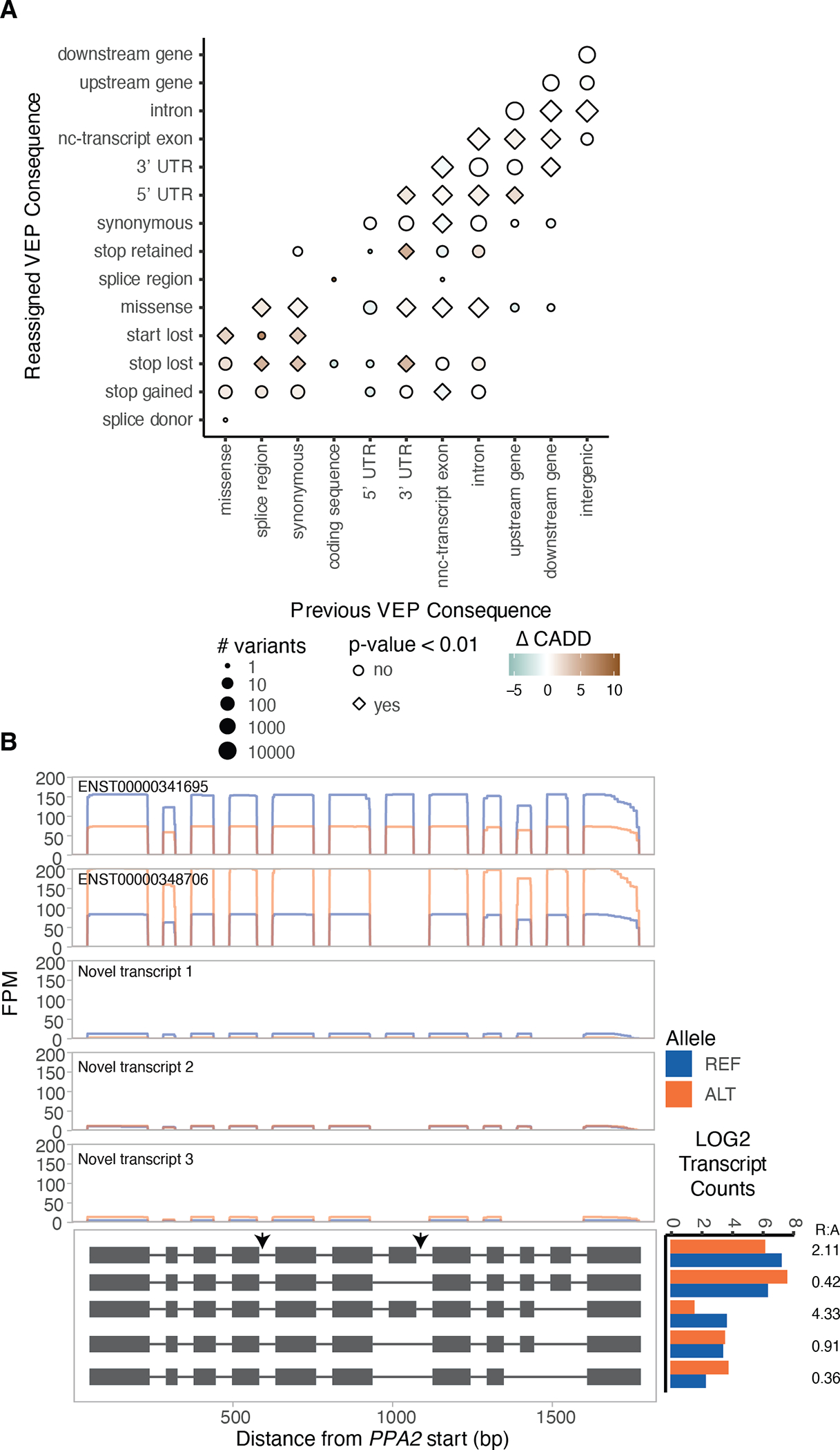
Regulation of transcript structure generates transcript diversity and plays an important role in human disease1-7. The advent of long-read sequencing technologies offers the opportunity to study the role of genetic variation in transcript structure8-16. In this Article, we present a large human long-read RNA-seq dataset using the Oxford Nanopore Technologies platform from 88 samples from Genotype-Tissue Expression (GTEx) tissues and cell lines, complementing the GTEx resource. We identified just over 70,000 novel transcripts for annotated genes, and validated the protein expression of 10% of novel transcripts. We developed a new computational package, LORALS, to analyse the genetic effects of rare and common variants on the transcriptome by allele-specific analysis of long reads. We characterized allele-specific expression and transcript structure events, providing new insights into the specific transcript alterations caused by common and rare genetic variants and highlighting the resolution gained from long-read data. We were able to perturb the transcript structure upon knockdown of PTBP1, an RNA binding protein that mediates splicing, thereby finding genetic regulatory effects that are modified by the cellular environment. Finally, we used this dataset to enhance variant interpretation and study rare variants leading to aberrant splicing patterns.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures
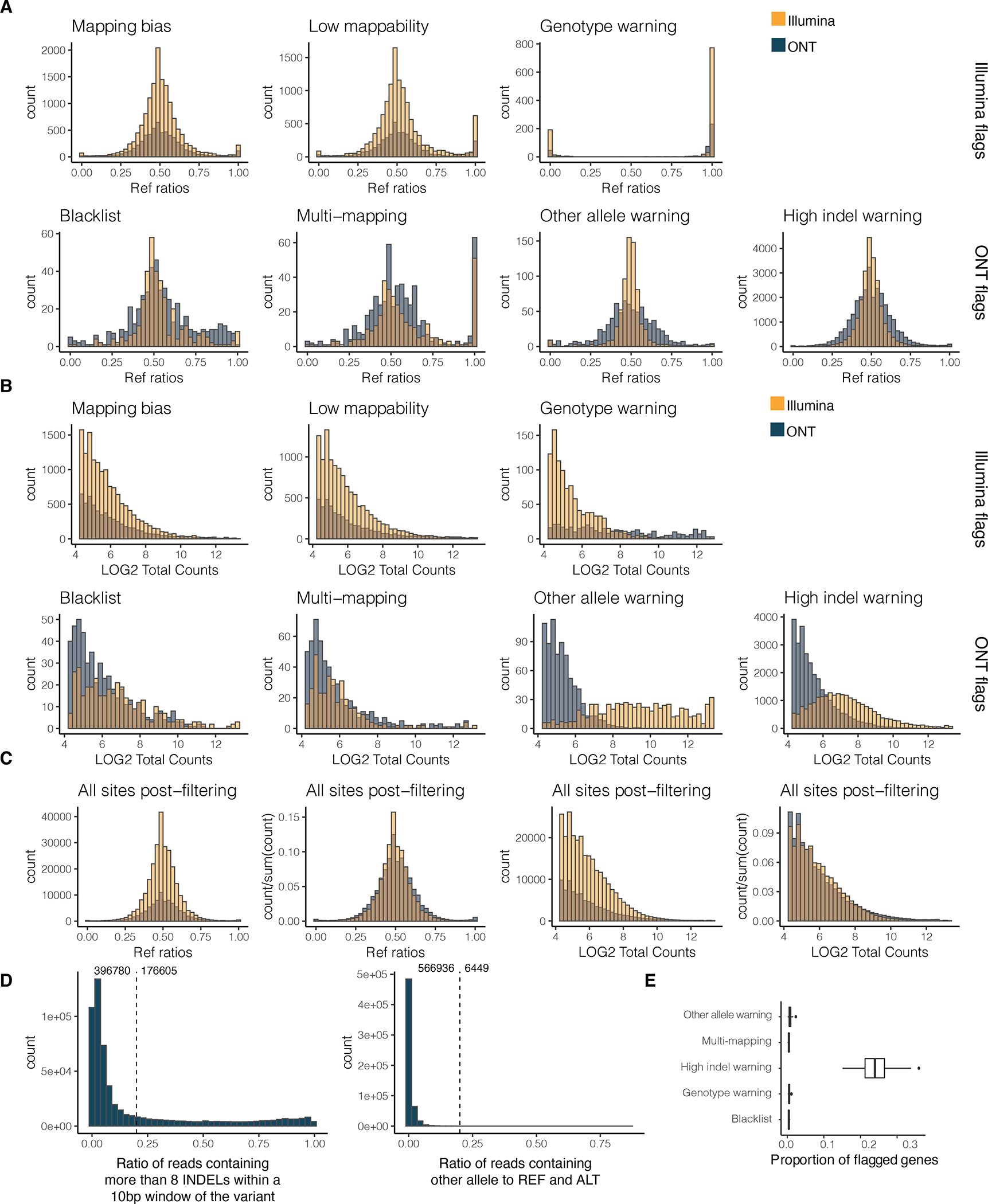
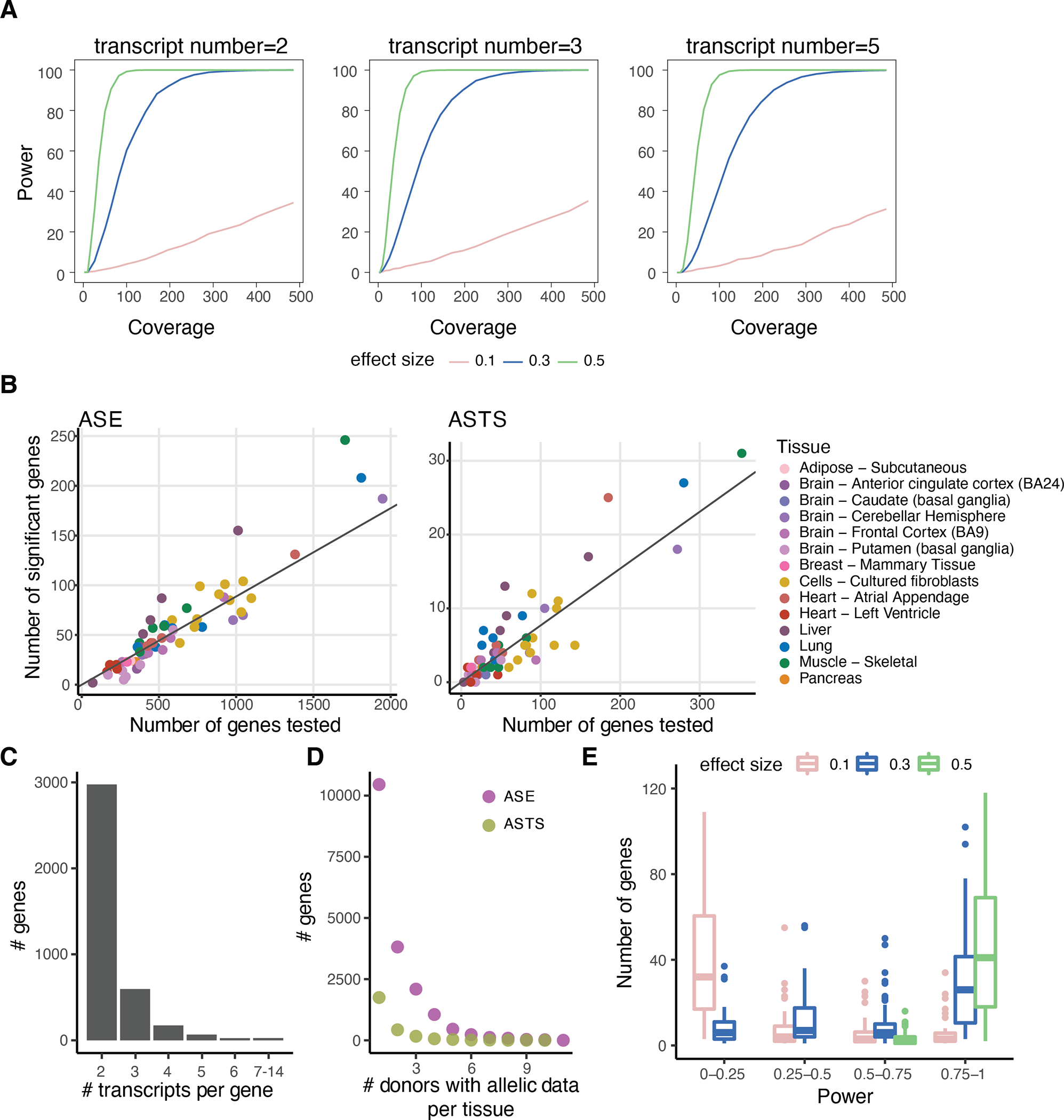
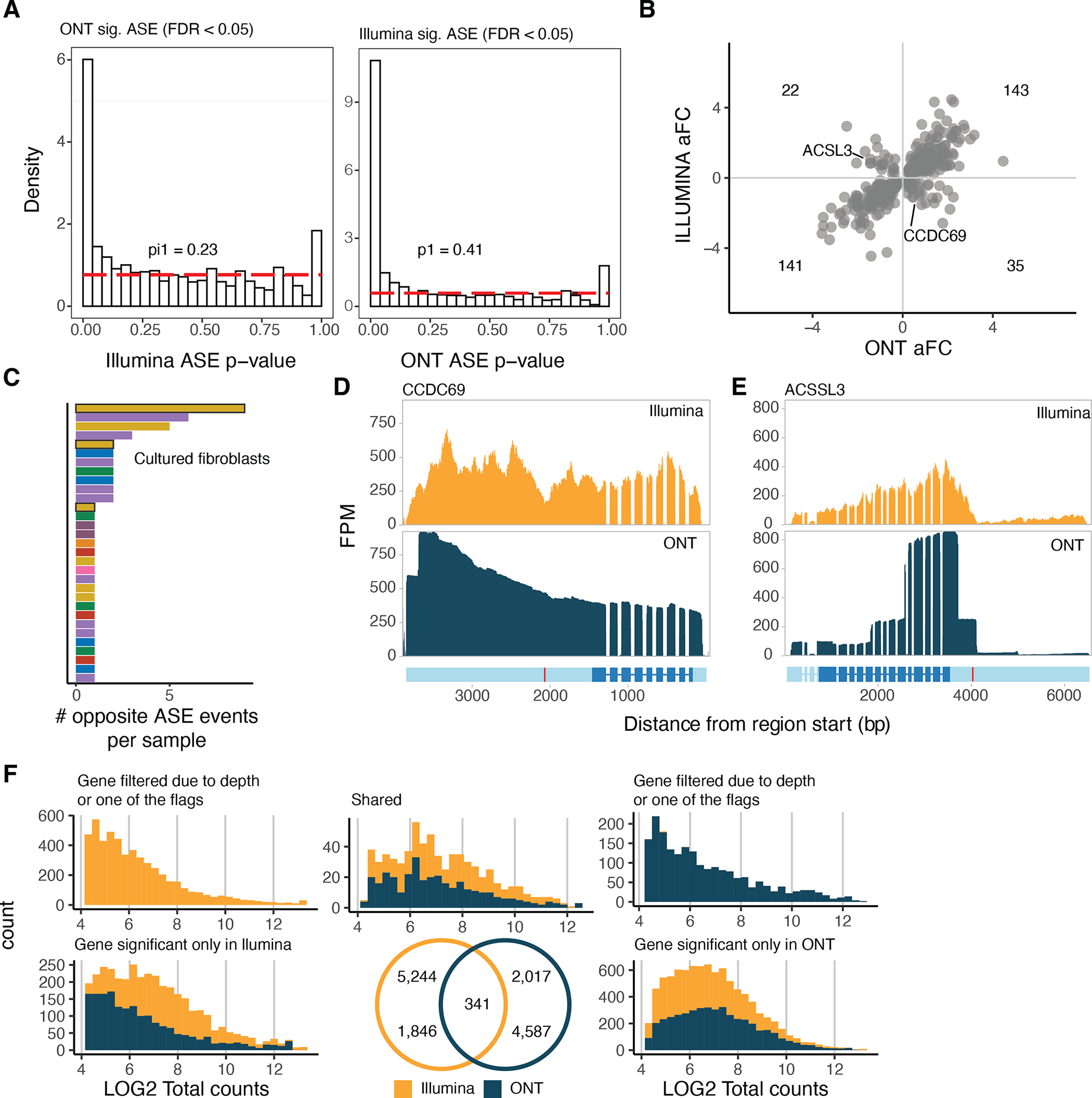
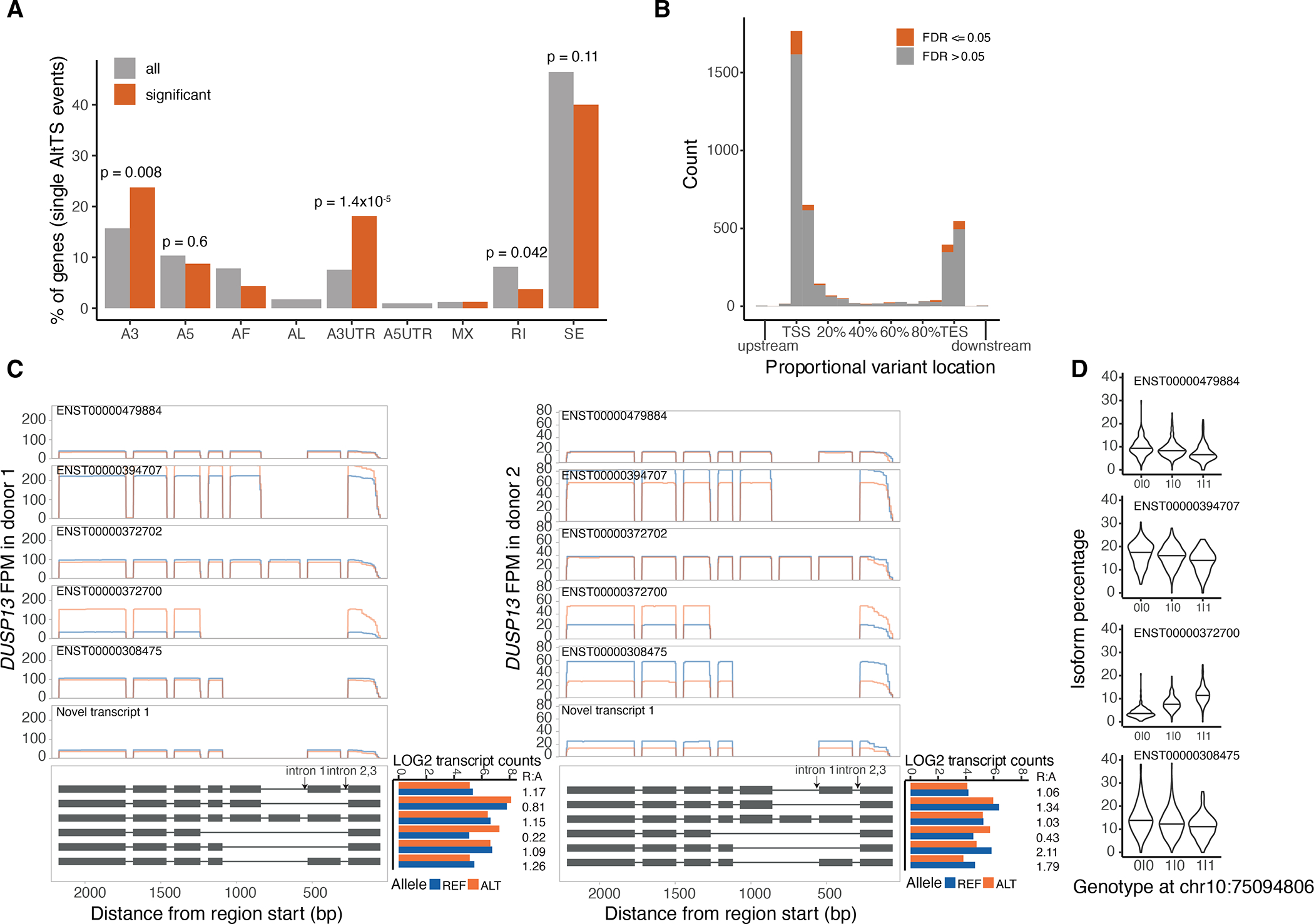
References
Methods References
-
- Alasoo K Wiggleplotr: Make read coverage plots from bigwig files. (2017).
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
