

Architecture and self-assembly of the jumbo bacteriophage nuclear shell
- PMID: 35922510
- PMCID: PMC9365700
- DOI: 10.1038/s41586-022-05013-4
Architecture and self-assembly of the jumbo bacteriophage nuclear shell
Abstract
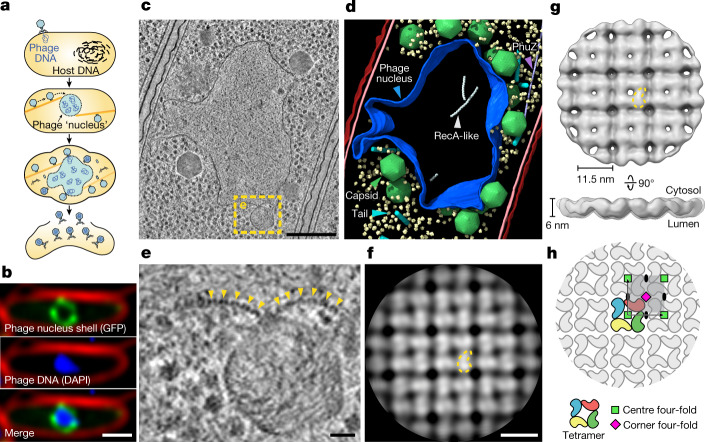
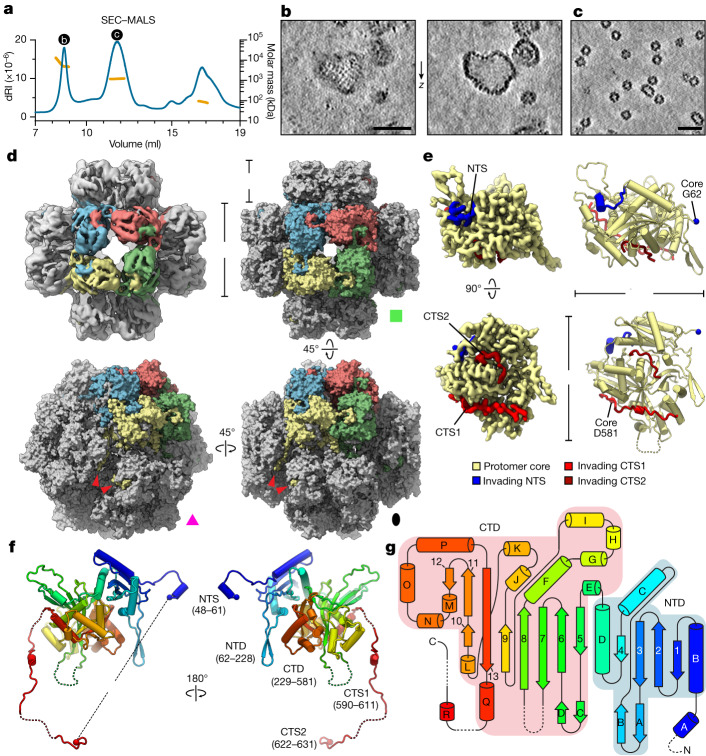
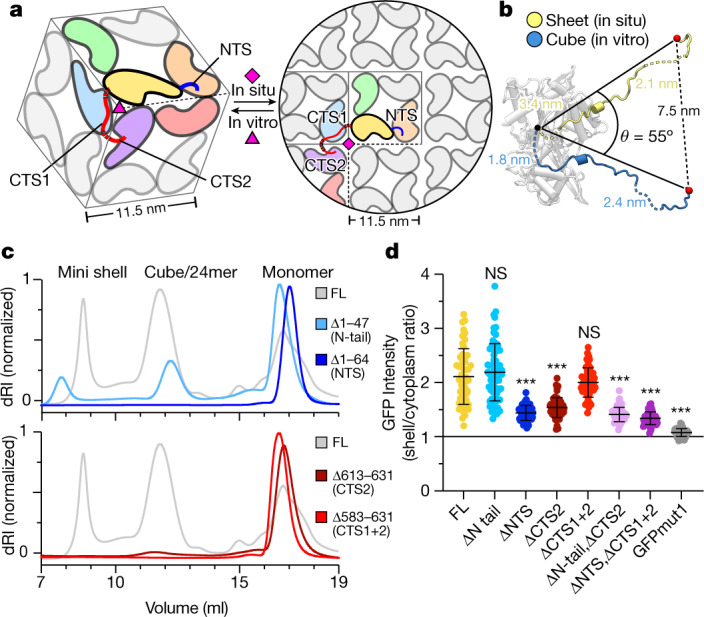
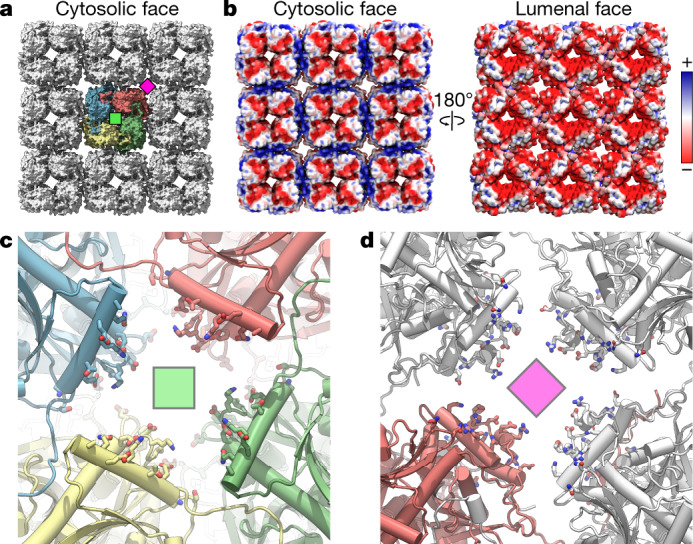
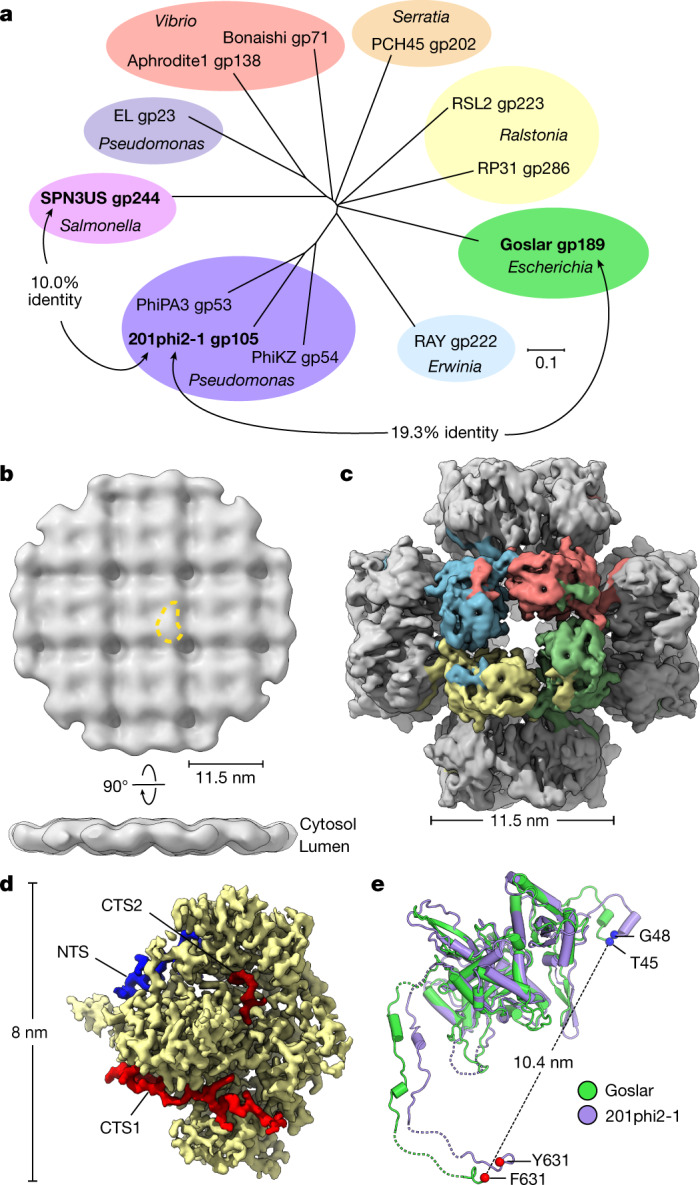
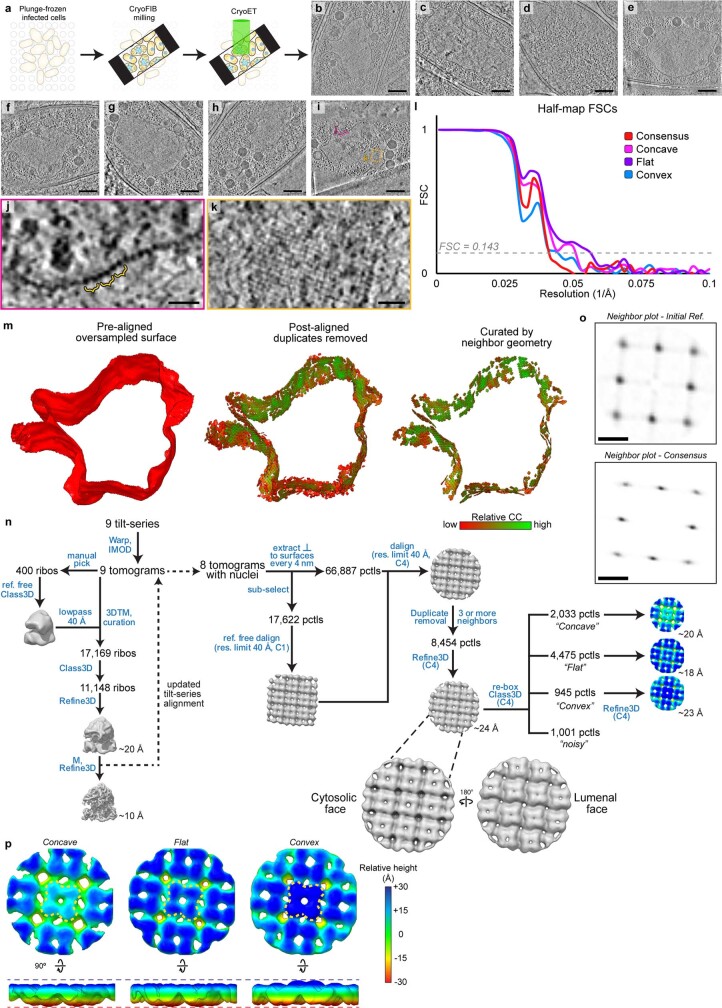
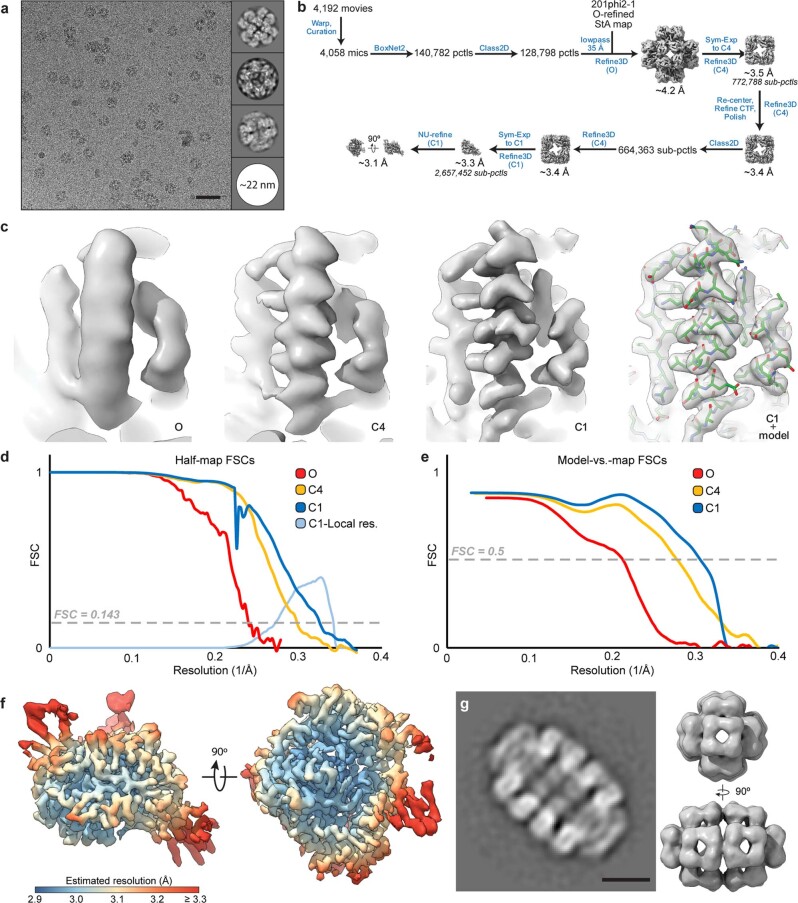
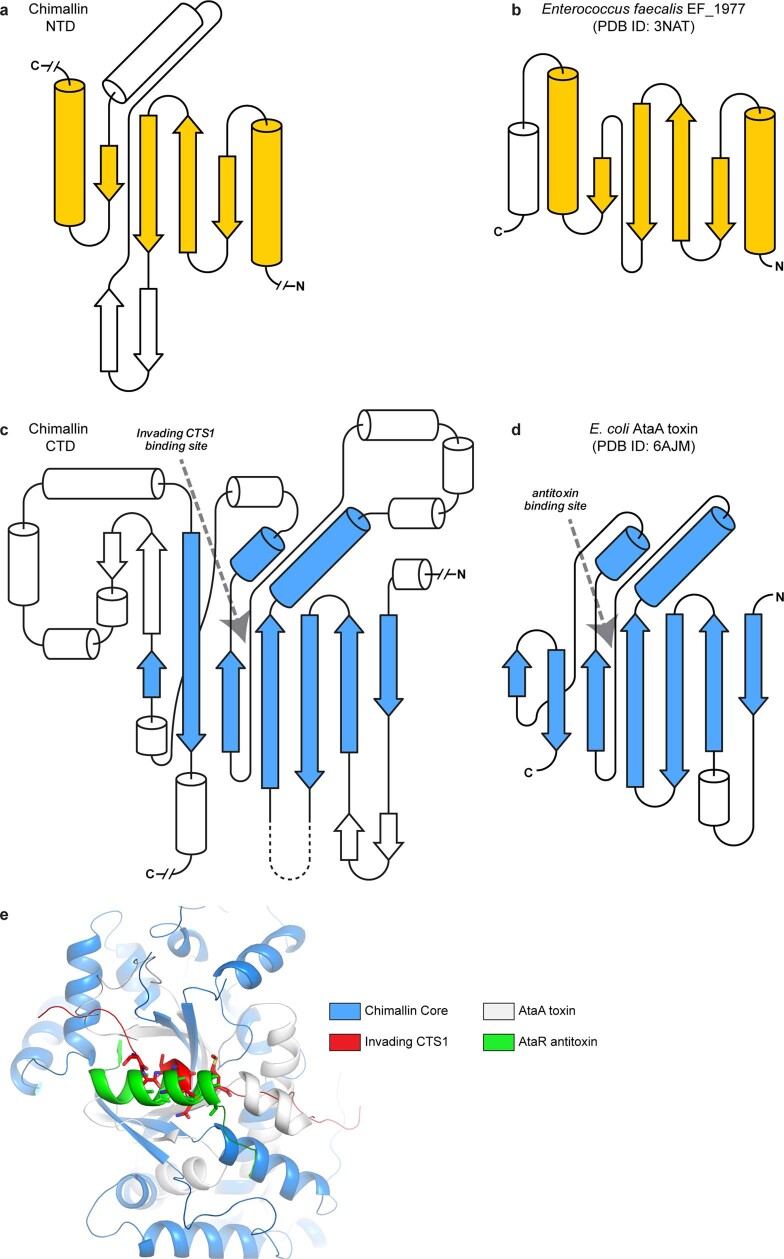
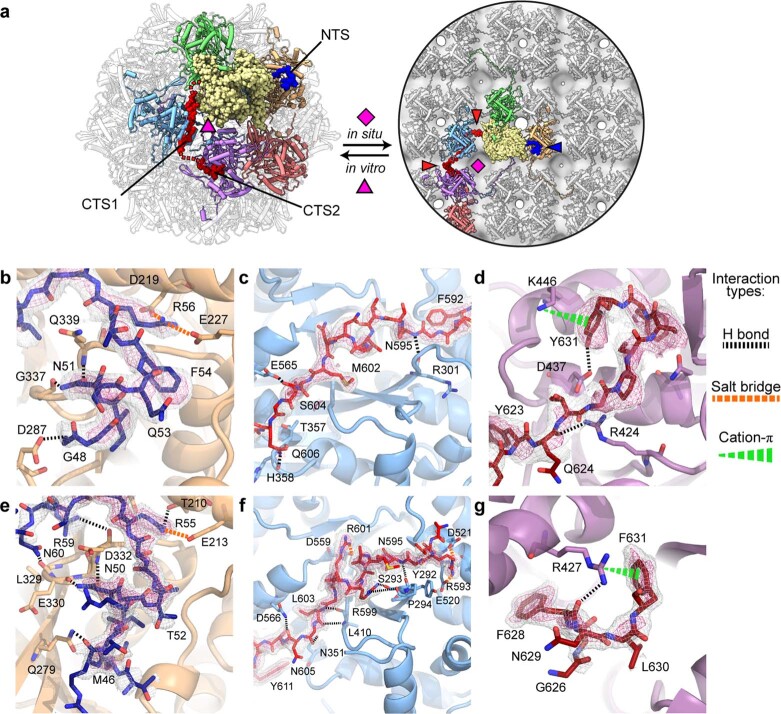
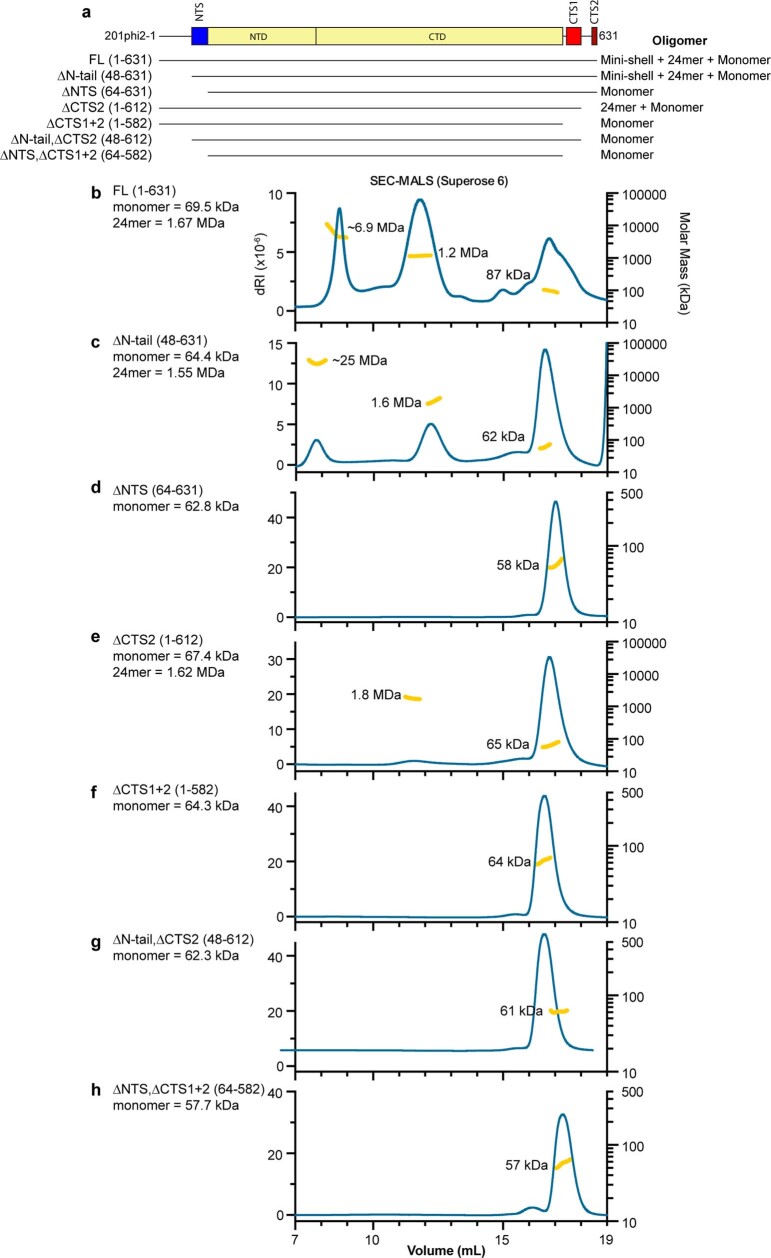
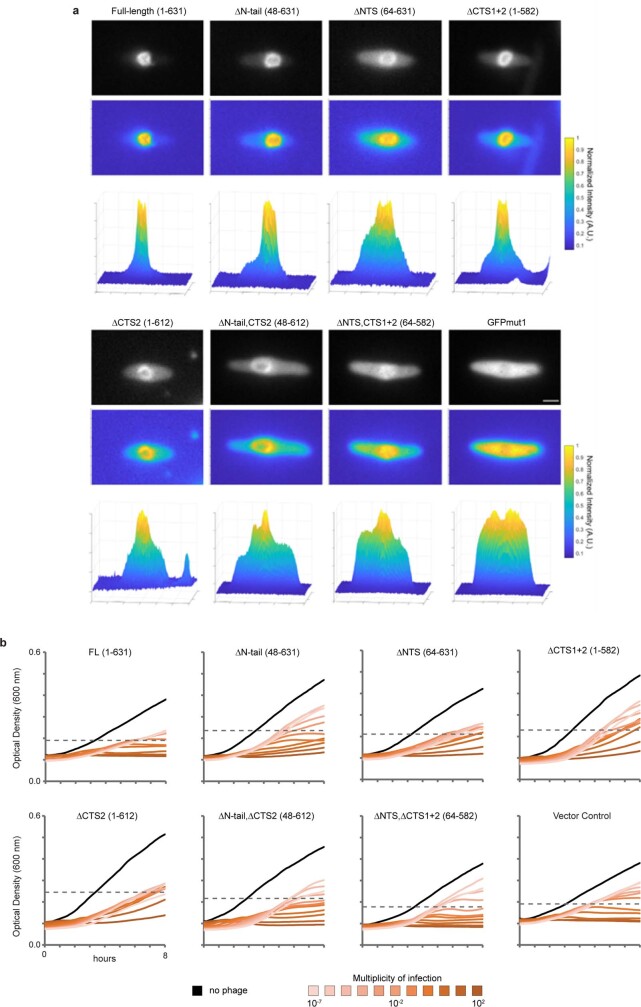
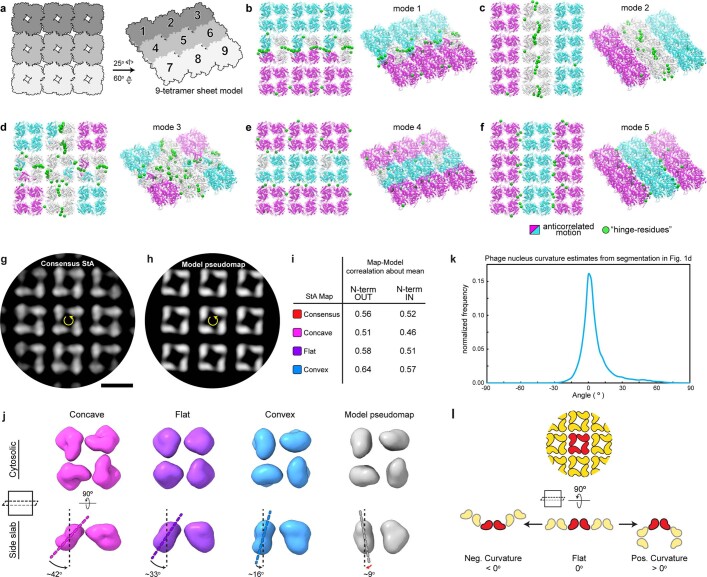
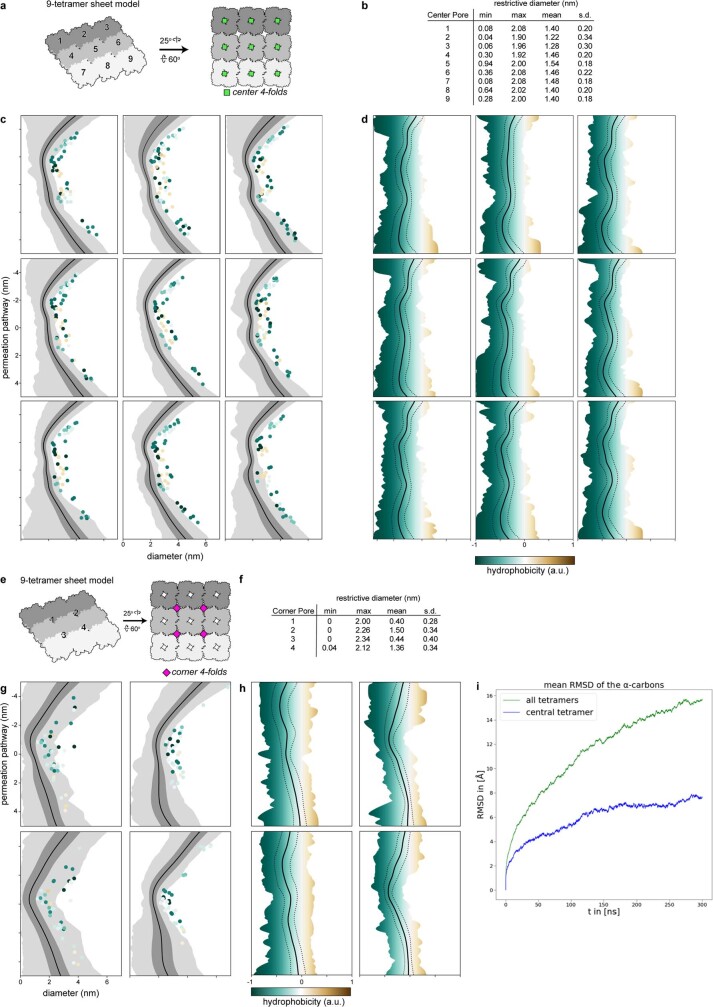
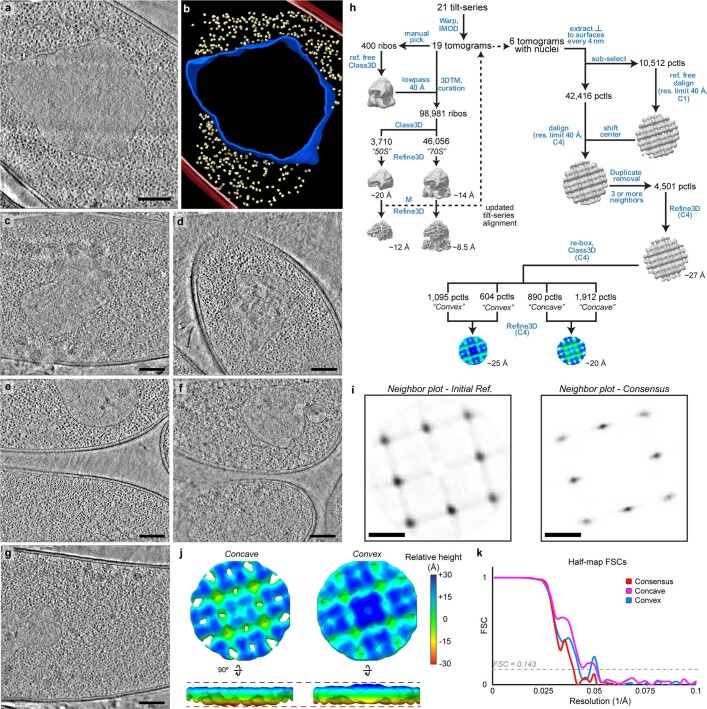
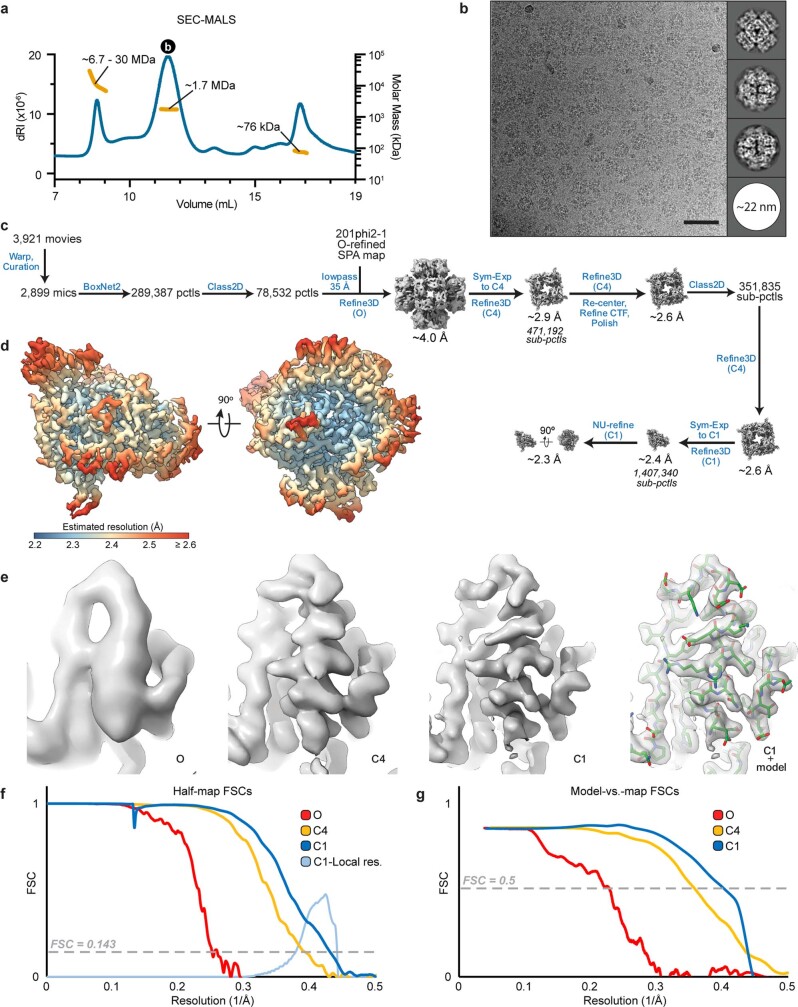
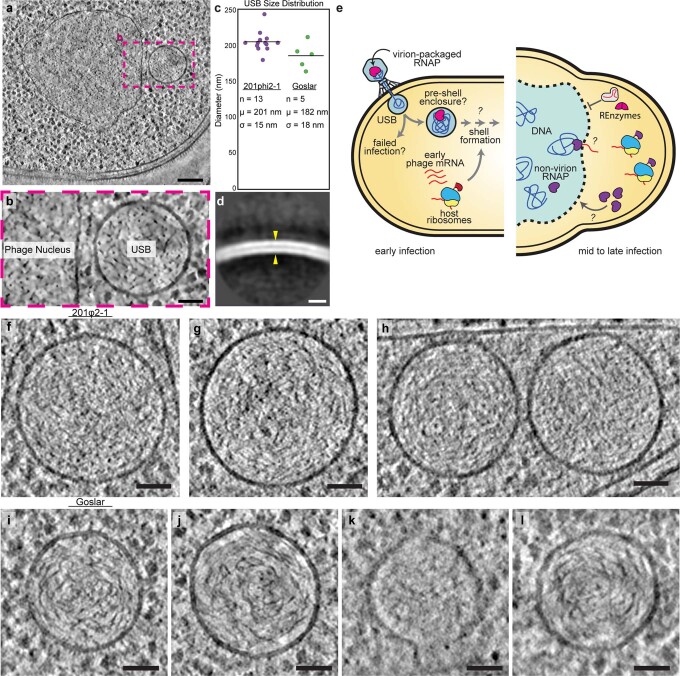
Bacteria encode myriad defences that target the genomes of infecting bacteriophage, including restriction-modification and CRISPR-Cas systems1. In response, one family of large bacteriophages uses a nucleus-like compartment to protect its replicating genomes by excluding host defence factors2-4. However, the principal composition and structure of this compartment remain unknown. Here we find that the bacteriophage nuclear shell assembles primarily from one protein, which we name chimallin (ChmA). Combining cryo-electron tomography of nuclear shells in bacteriophage-infected cells and cryo-electron microscopy of a minimal chimallin compartment in vitro, we show that chimallin self-assembles as a flexible sheet into closed micrometre-scale compartments. The architecture and assembly dynamics of the chimallin shell suggest mechanisms for its nucleation and growth, and its role as a scaffold for phage-encoded factors mediating macromolecular transport, cytoskeletal interactions, and viral maturation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
