

Cellular senescence: the good, the bad and the unknown
- PMID: 35922662
- PMCID: PMC9362342
- DOI: 10.1038/s41581-022-00601-z
Cellular senescence: the good, the bad and the unknown
Abstract
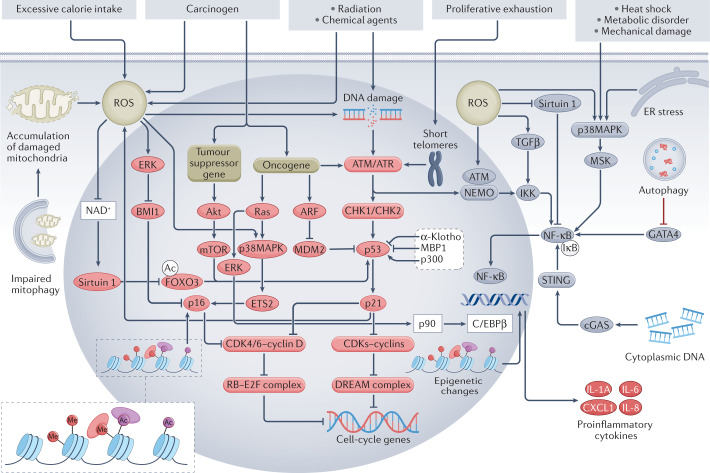
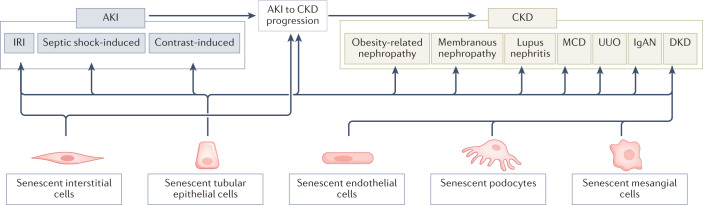
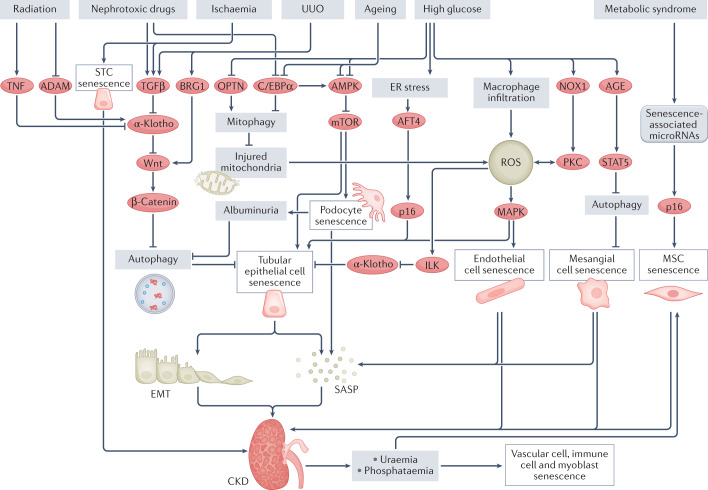
Cellular senescence is a ubiquitous process with roles in tissue remodelling, including wound repair and embryogenesis. However, prolonged senescence can be maladaptive, leading to cancer development and age-related diseases. Cellular senescence involves cell-cycle arrest and the release of inflammatory cytokines with autocrine, paracrine and endocrine activities. Senescent cells also exhibit morphological alterations, including flattened cell bodies, vacuolization and granularity in the cytoplasm and abnormal organelles. Several biomarkers of cellular senescence have been identified, including SA-βgal, p16 and p21; however, few markers have high sensitivity and specificity. In addition to driving ageing, senescence of immune and parenchymal cells contributes to the development of a variety of diseases and metabolic disorders. In the kidney, senescence might have beneficial roles during development and recovery from injury, but can also contribute to the progression of acute kidney injury and chronic kidney disease. Therapies that target senescence, including senolytic and senomorphic drugs, stem cell therapies and other interventions, have been shown to extend lifespan and reduce tissue injury in various animal models. Early clinical trials confirm that senotherapeutic approaches could be beneficial in human disease. However, larger clinical trials are needed to translate these approaches to patient care.
© 2022. Springer Nature Limited.
Conflict of interest statement
L.O.L. is an adviser to AstraZeneca, CureSpec, Beren, Ribocure Pharmaceuticals and Butterfly Biosciences. Patents on senolytic drugs and their uses are held by the Mayo Clinic. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies. The other authors declare no conflicts of interest.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
