

Genes expressed at low levels raise false discovery rates in RNA samples contaminated with genomic DNA
- PMID: 35922750
- PMCID: PMC9351092
- DOI: 10.1186/s12864-022-08785-1
Genes expressed at low levels raise false discovery rates in RNA samples contaminated with genomic DNA
Abstract
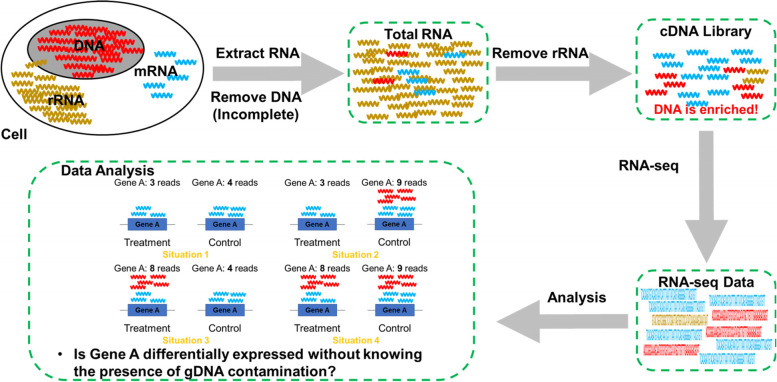
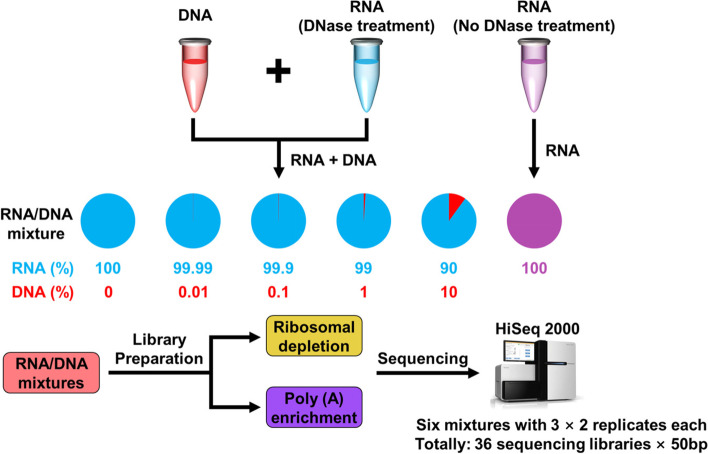
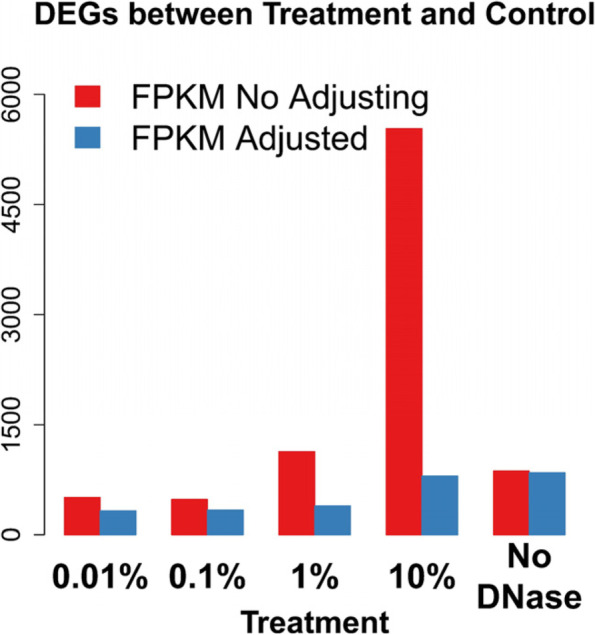
Background: RNA preparations contaminated with genomic DNA (gDNA) are frequently disregarded by RNA-seq studies. Such contamination may generate false results; however, their effect on the outcomes of RNA-seq analyses is unknown. To address this gap in our knowledge, here we added different concentrations of gDNA to total RNA preparations and subjected them to RNA-seq analysis.
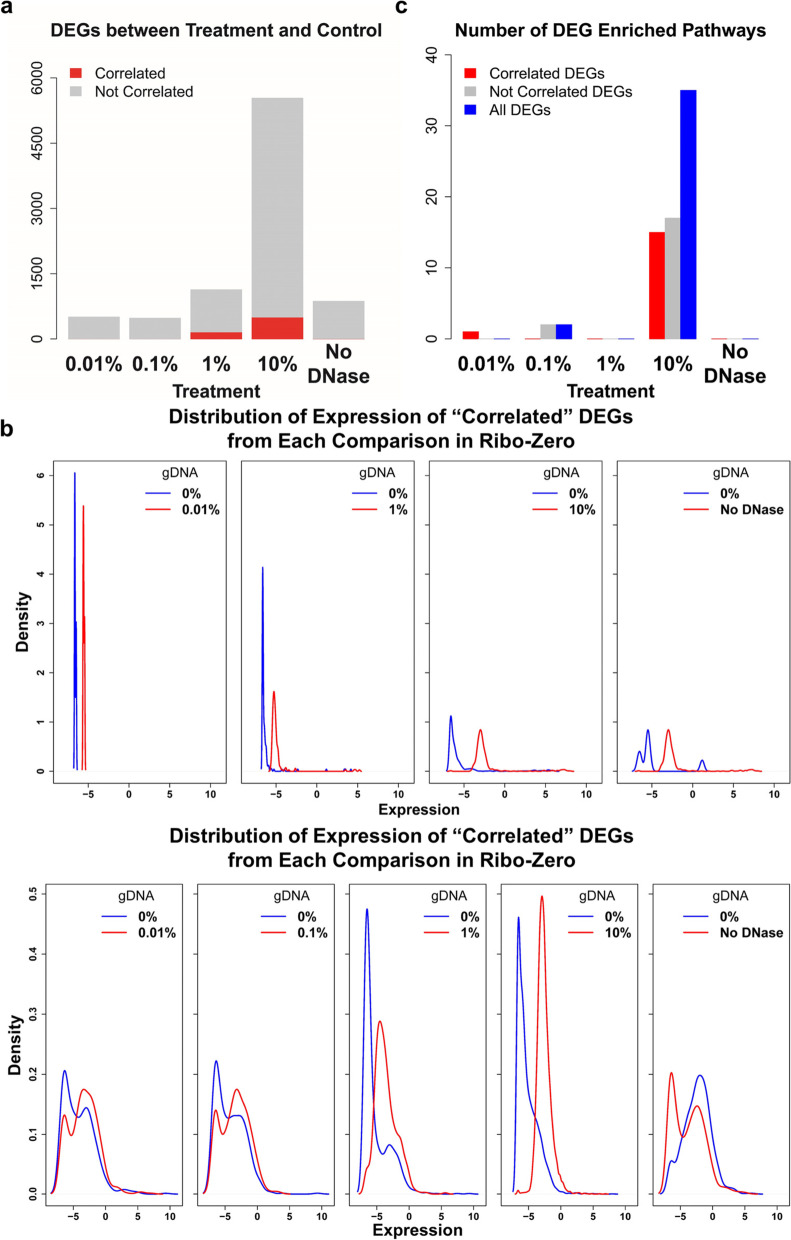
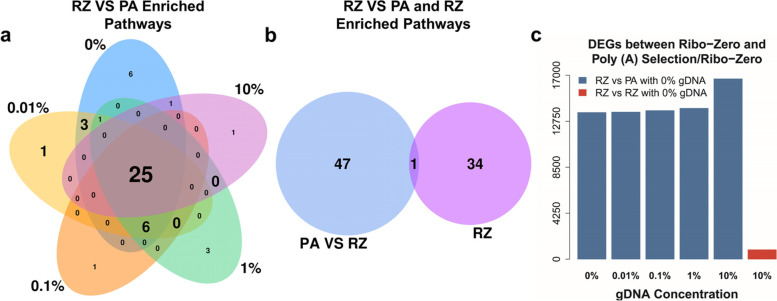
Results: We found that the contaminating gDNA altered the quantification of transcripts at relatively high concentrations. Differentially expressed genes (DEGs) resulting from gDNA contamination may therefore contribute to higher rates of false enrichment of pathways compared with analogous samples lacking numerous DEGs. A strategy was developed to correct gene expression levels in gDNA-contaminated RNA samples, which assessed the magnitude of contamination to improve the reliability of the results.
Conclusions: Our study indicates that caution must be exercised when interpreting results associated with low-abundance transcripts. The data provided here will likely serve as a valuable resource to evaluate the influence of gDNA contamination on RNA-seq analysis, particularly related to the detection of putative novel gene elements.
Keywords: False Discoveries; Genomic DNA Contamination; RNA-seq.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

Similar articles
-
CleanUpRNAseq: An R/Bioconductor Package for Detecting and Correcting DNA Contamination in RNA-Seq Data.BioTech (Basel). 2024 Aug 3;13(3):30. doi: 10.3390/biotech13030030. BioTech (Basel). 2024. PMID: 39189209 Free PMC article.
-
Assessment of DNA Contamination in RNA Samples Based on Ribosomal DNA.J Vis Exp. 2018 Jan 22;(131):55451. doi: 10.3791/55451. J Vis Exp. 2018. PMID: 29443017 Free PMC article.
-
Development of a versatile TaqMan™ real-time quantitative PCR (RT-qPCR) compliant anchor sequence to quantify bacterial gene transcripts from RNA samples containing carryover genomic DNA.BMC Biotechnol. 2013 Jan 31;13:7. doi: 10.1186/1472-6750-13-7. BMC Biotechnol. 2013. PMID: 23369378 Free PMC article.
-
Correction of RT-qPCR data for genomic DNA-derived signals with ValidPrime.Nucleic Acids Res. 2012 Apr;40(7):e51. doi: 10.1093/nar/gkr1259. Epub 2012 Jan 6. Nucleic Acids Res. 2012. PMID: 22228834 Free PMC article.
-
Uncovering the complexity of transcriptomes with RNA-Seq.J Biomed Biotechnol. 2010;2010:853916. doi: 10.1155/2010/853916. Epub 2010 Jun 27. J Biomed Biotechnol. 2010. PMID: 20625424 Free PMC article. Review.
Cited by
-
CleanUpRNAseq: An R/Bioconductor Package for Detecting and Correcting DNA Contamination in RNA-Seq Data.BioTech (Basel). 2024 Aug 3;13(3):30. doi: 10.3390/biotech13030030. BioTech (Basel). 2024. PMID: 39189209 Free PMC article.
-
Serum RNA Profile Reflects Fluid Status and Atrophic Retinal Changes in Neovascular Age-Related Macular Degeneration.Int J Mol Sci. 2025 May 19;26(10):4852. doi: 10.3390/ijms26104852. Int J Mol Sci. 2025. PMID: 40429992 Free PMC article.
-
Efficient profiling of total RNA in single cells with STORM-seq.bioRxiv [Preprint]. 2025 May 20:2022.03.14.484332. doi: 10.1101/2022.03.14.484332. bioRxiv. 2025. PMID: 40475517 Free PMC article. Preprint.
-
Assessing and mitigating batch effects in large-scale omics studies.Genome Biol. 2024 Oct 3;25(1):254. doi: 10.1186/s13059-024-03401-9. Genome Biol. 2024. PMID: 39363244 Free PMC article. Review.
-
A single workflow for multi-species blood transcriptomics.BMC Genomics. 2024 Mar 16;25(1):282. doi: 10.1186/s12864-024-10208-2. BMC Genomics. 2024. PMID: 38493105 Free PMC article.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
