

Identifying novel host-based diagnostic biomarker panels for COVID-19: a whole-blood/nasopharyngeal transcriptome meta-analysis
- PMID: 35922752
- PMCID: PMC9347150
- DOI: 10.1186/s10020-022-00513-5
Identifying novel host-based diagnostic biomarker panels for COVID-19: a whole-blood/nasopharyngeal transcriptome meta-analysis
Abstract
Background: Regardless of improvements in controlling the COVID-19 pandemic, the lack of comprehensive insight into SARS-COV-2 pathogenesis is still a sophisticated challenge. In order to deal with this challenge, we utilized advanced bioinformatics and machine learning algorithms to reveal more characteristics of SARS-COV-2 pathogenesis and introduce novel host response-based diagnostic biomarker panels.
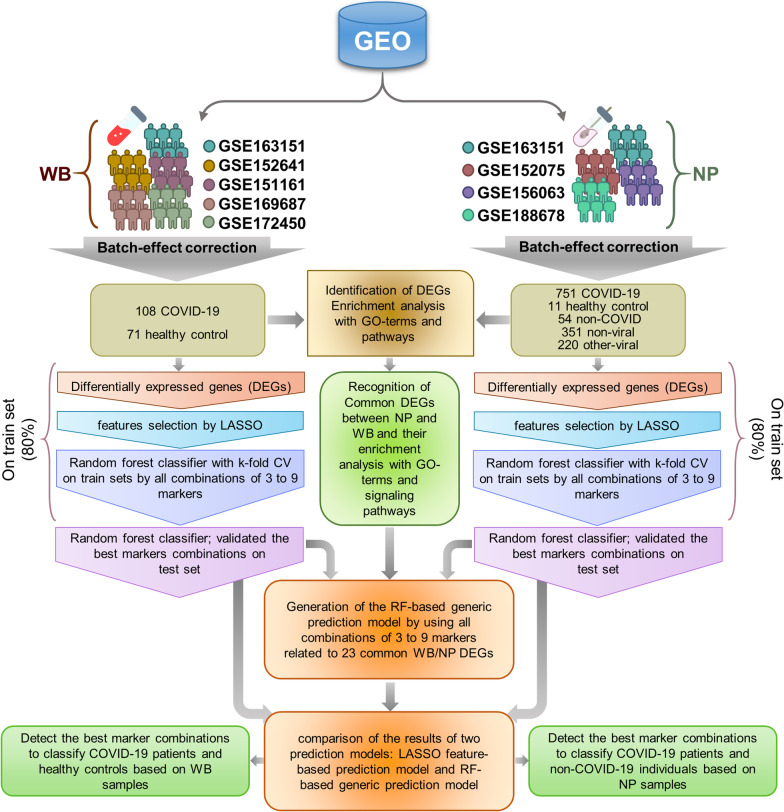
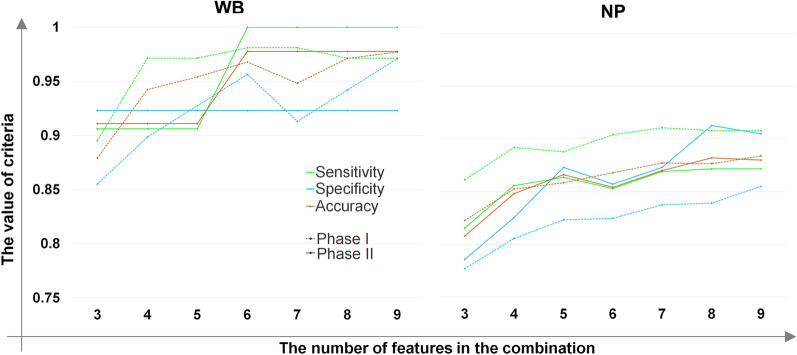
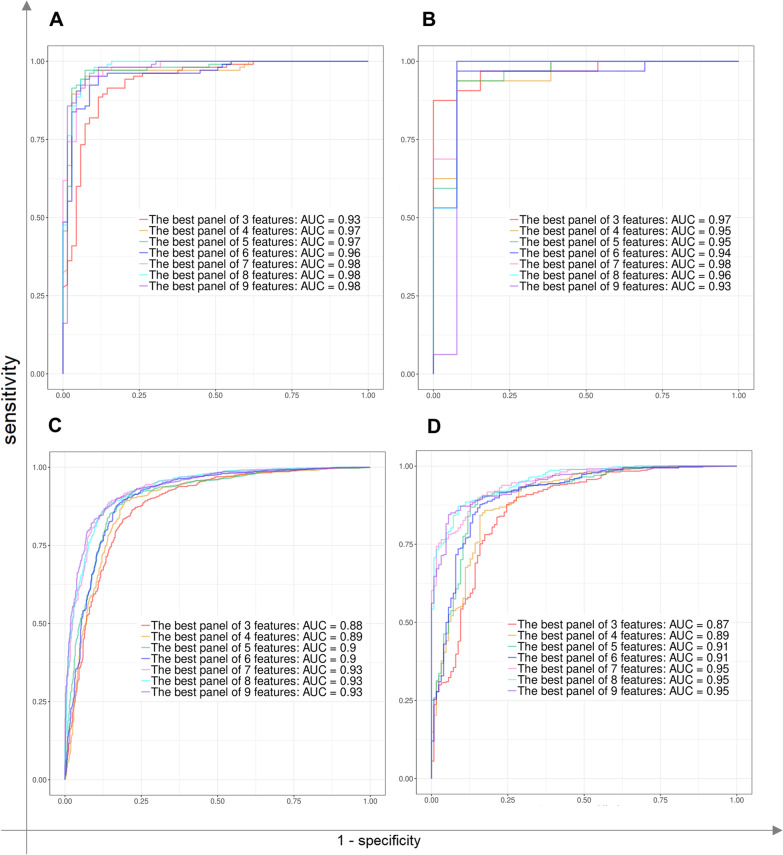
Methods: In the present study, eight published RNA-Seq datasets related to whole-blood (WB) and nasopharyngeal (NP) swab samples of patients with COVID-19, other viral and non-viral acute respiratory illnesses (ARIs), and healthy controls (HCs) were integrated. To define COVID-19 meta-signatures, Gene Ontology and pathway enrichment analyses were applied to compare COVID-19 with other similar diseases. Additionally, CIBERSORTx was executed in WB samples to detect the immune cell landscape. Furthermore, the optimum WB- and NP-based diagnostic biomarkers were identified via all the combinations of 3 to 9 selected features and the 2-phases machine learning (ML) method which implemented k-fold cross validation and independent test set validation.
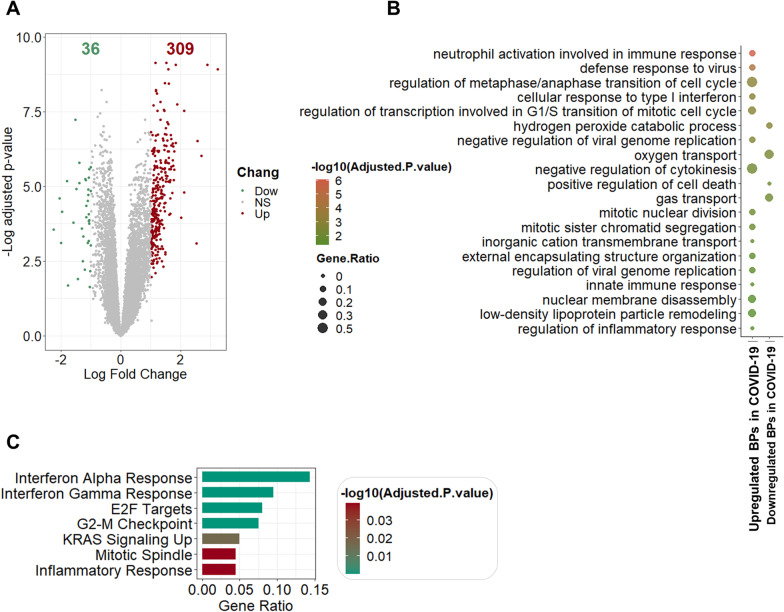
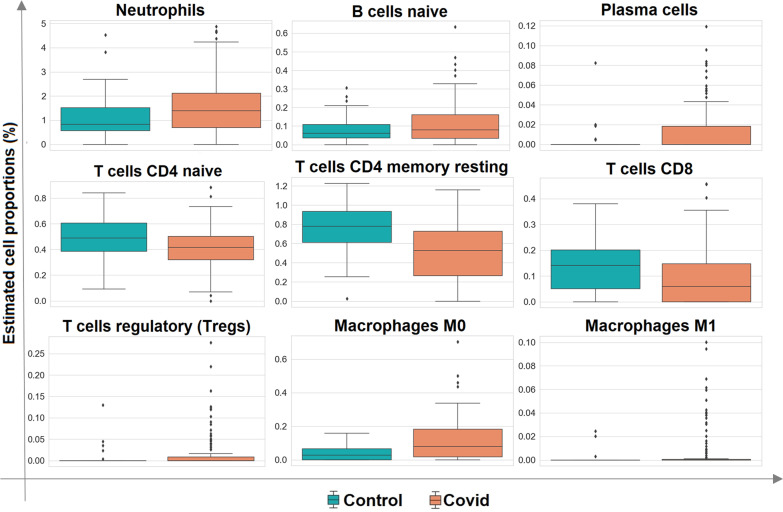
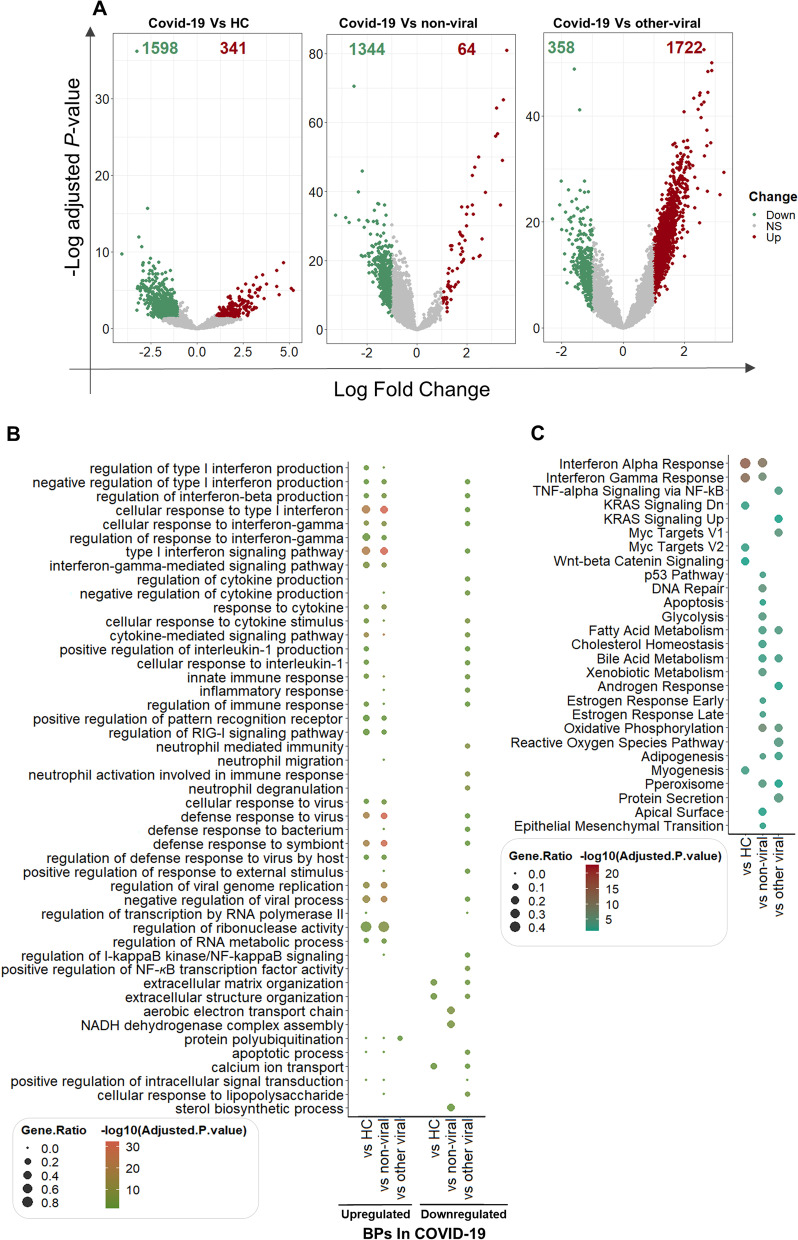
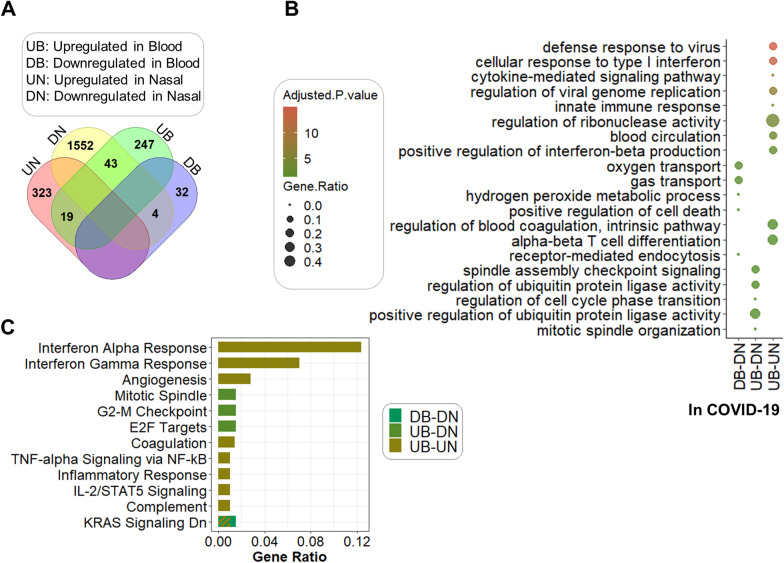
Results: The host gene meta-signatures obtained for SARS-COV-2 infection were different in the WB and NP samples. The gene ontology and enrichment results of the WB dataset represented the enhancement in inflammatory host response, cell cycle, and interferon signature in COVID-19 patients. Furthermore, NP samples of COVID-19 in comparison with HC and non-viral ARIs showed the significant upregulation of genes associated with cytokine production and defense response to the virus. In contrast, these pathways in COVID-19 compared to other viral ARIs were strikingly attenuated. Notably, immune cell proportions of WB samples altered in COVID-19 versus HC. Moreover, the optimum WB- and NP-based diagnostic panels after two phases of ML-based validation included 6 and 8 markers with an accuracy of 97% and 88%, respectively.
Conclusions: Based on the distinct gene expression profiles of WB and NP, our results indicated that SARS-COV-2 function is body-site-specific, although according to the common signature in WB and NP COVID-19 samples versus controls, this virus also induces a global and systematic host response to some extent. We also introduced and validated WB- and NP-based diagnostic biomarkers using ML methods which can be applied as a complementary tool to diagnose the COVID-19 infection from non-COVID cases.
Keywords: Biomarker; COVID-19; Data integration; Nasopharyngeal swab; Pathogenesis; Random forest; SARS-COV-2; Systems biology; Whole blood.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
