

Identification and Characterization of Two Novel Compounds: Heterozygous Variants of Lipoprotein Lipase in Two Pedigrees With Type I Hyperlipoproteinemia
- PMID: 35923617
- PMCID: PMC9339609
- DOI: 10.3389/fendo.2022.874608
Identification and Characterization of Two Novel Compounds: Heterozygous Variants of Lipoprotein Lipase in Two Pedigrees With Type I Hyperlipoproteinemia
Abstract
Background: Type I hyperlipoproteinemia, characterized by severe hypertriglyceridemia, is caused mainly by loss-of-function mutation of the lipoprotein lipase (LPL) gene. To date, more than 200 mutations in the LPL gene have been reported, while only a limited number of mutations have been evaluated for pathogenesis.
Objective: This study aims to explore the molecular mechanisms underlying lipoprotein lipase deficiency in two pedigrees with type 1 hyperlipoproteinemia.
Methods: We conducted a systematic clinical and genetic analysis of two pedigrees with type 1 hyperlipoproteinemia. Postheparin plasma of all the members was used for the LPL activity analysis. In vitro studies were performed in HEK-293T cells that were transiently transfected with wild-type or variant LPL plasmids. Furthermore, the production and activity of LPL were analyzed in cell lysates or culture medium.
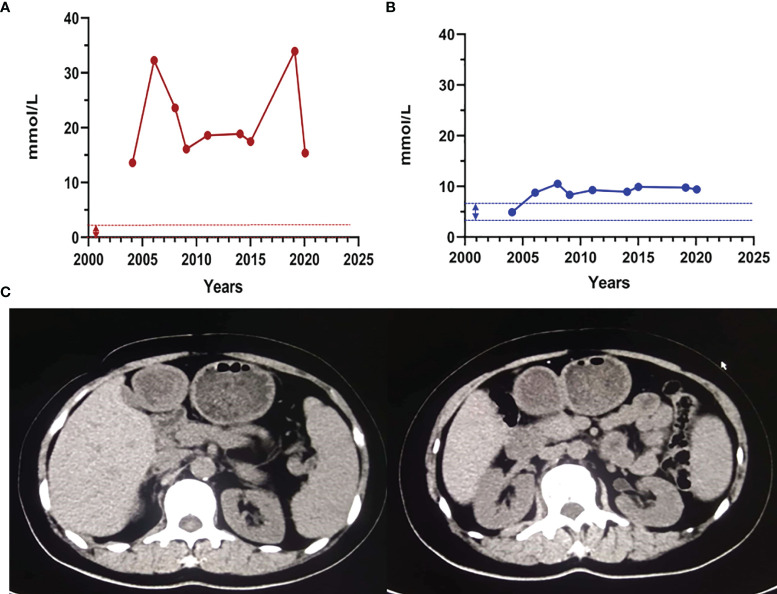
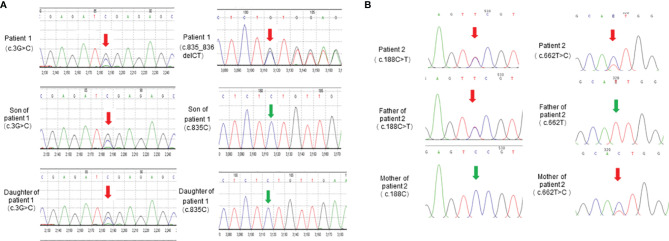
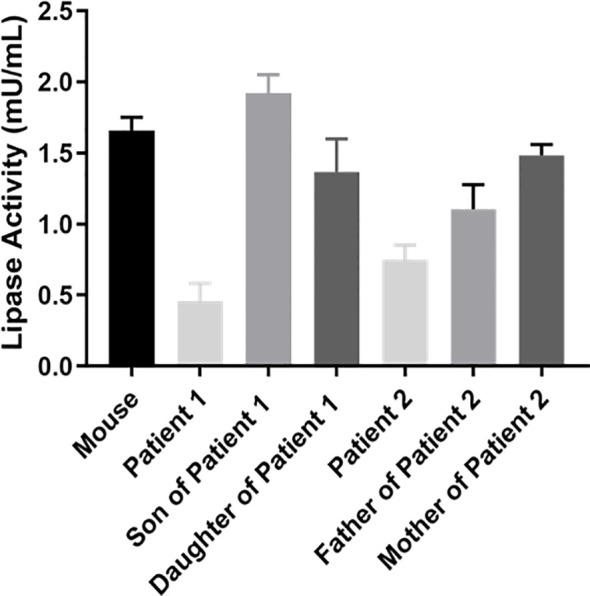
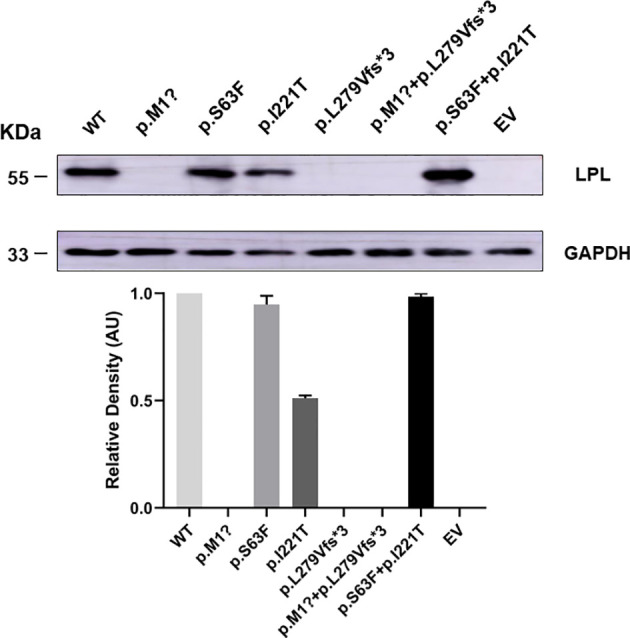
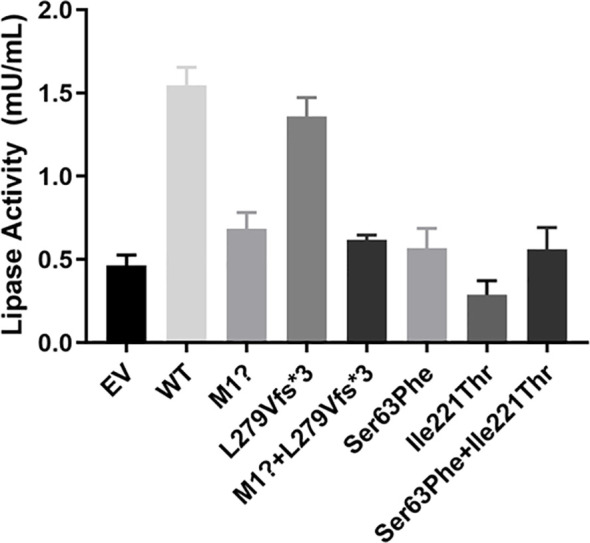
Results: Proband 1 developed acute pancreatitis in youth, and her serum triglycerides (TGs) continued to be at an ultrahigh level, despite the application of various lipid-lowering drugs. Proband 2 was diagnosed with type 1 hyperlipoproteinemia at 9 months of age, and his serum TG levels were mildly elevated with treatment. Two novel compound heterozygous variants of LPL (c.3G>C, p. M1? and c.835_836delCT, p. L279Vfs*3, c.188C>T, p. Ser63Phe and c.662T>C, p. Ile221Thr) were identified in the two probands. The postheparin LPL activity of probands 1 and 2 showed decreases of 72.22 ± 9.46% (p<0.01) and 54.60 ± 9.03% (p<0.01), respectively, compared with the control. In vitro studies showed a substantial reduction in the expression or enzyme activity of LPL in the LPL variants.
Conclusions: Two novel compound heterozygous variants of LPL induced defects in the expression and function of LPL and caused type I hyperlipoproteinemia. The functional characterization of these variants was in keeping with the postulated LPL mutant activity.
Keywords: hypertriglyceridemia; lipoprotein lipase (LPL); pedigree; type 1; variants.
Copyright © 2022 Wang, Cheng, Shi, Zhao, Gao, Fang, Jin, Han, Sun, Li, Zhao and Xu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
-
- Hoffmann MM, Jacob S, Luft D, Schmülling RM, Rett K, März W, et al. . Type I Hyperlipoproteinemia Due to a Novel Loss of Function Mutation of Lipoprotein Lipase, Cys(239)-->TRP, Associated with Recurrent Severe Pancreatitis. J Clin Endocrinol Metab (2000) 85(4):4795–8. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous