

Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: current and emerging role
- PMID: 35923930
- PMCID: PMC9340905
- DOI: 10.1177/17588359221113694
Next-generation selective estrogen receptor degraders and other novel endocrine therapies for management of metastatic hormone receptor-positive breast cancer: current and emerging role
Abstract
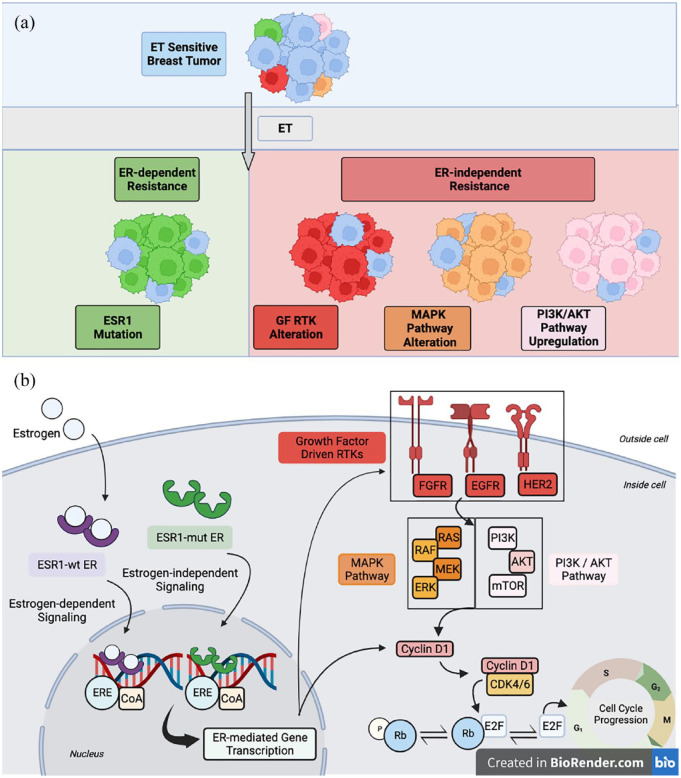
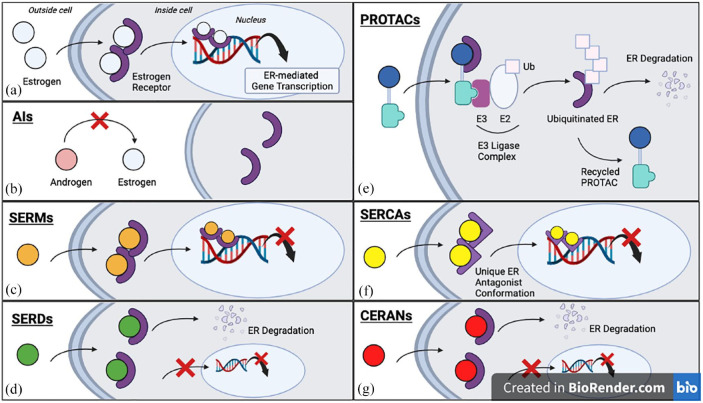
Endocrine therapy (ET) is a pivotal strategy to manage early- and advanced-stage estrogen receptor-positive (ER+) breast cancer. In patients with metastatic breast cancer (MBC), progression of disease inevitably occurs due to the presence of acquired or intrinsic resistance mechanisms. ET resistance can be driven by ligand-independent, ER-mediated signaling that promotes tumor proliferation in the absence of hormone, or ER-independent oncogenic signaling that circumvents endocrine regulated transcription pathways. Estrogen receptor 1 (ESR1) mutations induce constitutive ER activity and upregulate ER-dependent gene transcription, provoking resistance to estrogen deprivation and aromatase inhibitor therapy. The role ESR1 mutations play in regulating response to other therapies, such as the selective estrogen receptor degrader (SERD) fulvestrant and the available CDK4/6 inhibitors, is less clear. Novel oral SERDs and other next-generation ETs are in clinical development for ER+ breast cancer as single agents and in combination with established targeted therapies. Recent results from the phase III EMERALD trial demonstrated improved outcomes with the oral SERD elacestrant compared to standard anti-estrogen therapies in ER+ MBC after prior progression on ET, and other agents have shown promise in both the laboratory and early-phase clinical trials. In this review, we will discuss the emerging data related to oral SERDs and other novel ET in managing ER+ breast cancer. As clinical data continue to mature on these next-generation ETs, important questions will emerge related to the optimal sequence of therapeutic options and the genomic and molecular landscape of resistance to these agents.
Keywords: SERD; breast cancer; endocrine therapy; estrogen; metastatic disease.
© The Author(s), 2022.
Conflict of interest statement
Competing Interests: MRL: No disclosures. SAW: Consulting/Advisory Board – Foundation Medicine, Veracyte, Pfizer, Biovica, Hologic, Eli Lilly; Institutional Research Support – Genentech, Nuvation Bio, Regor Therapeutics, Eli Lilly. AB: Consultant/Advisory Board: Pfizer, Novartis, Genentech, Merck, Radius Health, Immunomedics/Gilead, Sanofi, Daiichi Pharma/Astra Zeneca, Phillips, Eli Lilly, Foundation Medicine; Contracted Research/Grant (to institution): Genentech, Novartis, Pfizer, Merck, Sanofi, Radius Health, Immunomedics/Gilead, Daiichi Pharma/Astra Zeneca, Eli Lilly. EH: Contracted Research/Grant (to institution): Abbvie, Acerta Pharma, Accutar Biotechnology, ADC Therapeutics, AKESOBIO Australia, Amgen, Aravive, ArQule, Artios, Arvinas, AstraZeneca, AtlasMedx, Black Diamond, Bliss BioPharmaceuticals, Boehringer Ingelheim, Cascadian Therapeutics, Clovis, Compugen, Cullen-Florentine, Curis, CytomX, Daiichi Sankyo, Dana Farber Cancer Inst, Dantari, Deciphera, Duality Biologics, eFFECTOR Therapeutics, Ellipses Pharma, Elucida Oncology, EMD Serono, Fochon, FujiFilm, G1 Therapeutics, H3 Biomedicine, Harpoon, Hutchinson MediPharma, Immunogen, Immunomedics, Incyte, Infinity Pharmaceuticals, InvestisBio, Jacobio, Karyopharm, Leap Therapeutics, Lilly, Lycera, Mabspace Biosciences, Macrogenics, MedImmune, Merck, Mersana, Merus, Millennium, Molecular Templates, Myriad Genetic Laboratories, Novartis, Nucana, Olema, OncoMed, Onconova Therapeutics, ORIC Pharmaceuticals, Orinove, Pfizer, Pharma Mar, Pieris Pharmaceuticals Pionyr, Immunotherapeutics, Plexxikon, Radius Health, Regeneron, Relay Therapeutics, Repertoire, Immune Medicine, Rgenix, Roche/Genentech, SeaGen, Sermonix Pharmaceuticals, Shattuck Labs, Silverback, StemCentRx, Sutro, Syndax, Syros, Taiho, TapImmune, Tesaro, Tolmar, Torque Therapeutics, Treadwell Therapeutics, Verastem, Vincerx Pharma, Zenith Epigenetics, Zymeworks; Consulting Advisory Role (all to institution only): Arcus, Arvinas, AstraZeneca, Black Diamond, Boehringer Ingelheim, CytomX, Daiichi Sankyo, Dantari, Deciphera Pharmaceuticals, Eisai, Greenwich LifeSciences, H3 Biomedicine iTeosJanssen, Lilly, Loxo, Merck, Mersana, Novartis, Orum Therapeutics, Pfizer, Propella Therapeutics, Puma Biotechnology, Relay Therapeutics, Roche/Genentech, SeaGen, Silverback Therapeutics. PR: Contracted Research/Grant (to institution): Grail/Illumina, Tempus, Novartis, AstraZeneca, Guardant, Epic Sciences, Inivata, Invitae/ArcherDx, Biotheranostics, Biovica, Foundation Medicine; Consultant/Advisory board: Novartis, AstraZeneca, Pfizer, Daiichi, Guardant, Natera, Inivata, Biovica, Epic Sciences, Tempus, Foundation Medicine.
Figures
References
-
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Aromatase inhibitors versus tamoxifen in early breast cancer: patient-level meta-analysis of the randomised trials. Lancet 2015; 386: 1341–1352. - PubMed
-
- DeSantis CE, Ma J, Gaudet MM, et al. Breast cancer statistics, 2019. CA Cancer J Clin 2019; 69: 438–451. - PubMed
-
- Brueggemeier RW. Aromatase inhibitors mechanisms of steroidal inhibitors. Breast Cancer Res Treat 1994; 30: 31–42. - PubMed
-
- Mauri D, Pavlidis N, Polyzos NP, et al. Survival with aromatase inhibitors and inactivators versus standard hormonal therapy in advanced breast cancer: meta-analysis. J Natl Cancer Inst 2006; 98: 1285–1291. - PubMed
-
- Gao JJ, Cheng J, Bloomquist E, et al. CDK4/6 inhibitor treatment for patients with hormone receptor-positive, HER2-negative, advanced or metastatic breast cancer: a US Food and Drug Administration pooled analysis. Lancet Oncol 2020; 21: 250–260. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
