

TREM2 Promotes Immune Evasion by Mycobacterium tuberculosis in Human Macrophages
- PMID: 35924849
- PMCID: PMC9426521
- DOI: 10.1128/mbio.01456-22
TREM2 Promotes Immune Evasion by Mycobacterium tuberculosis in Human Macrophages
Abstract
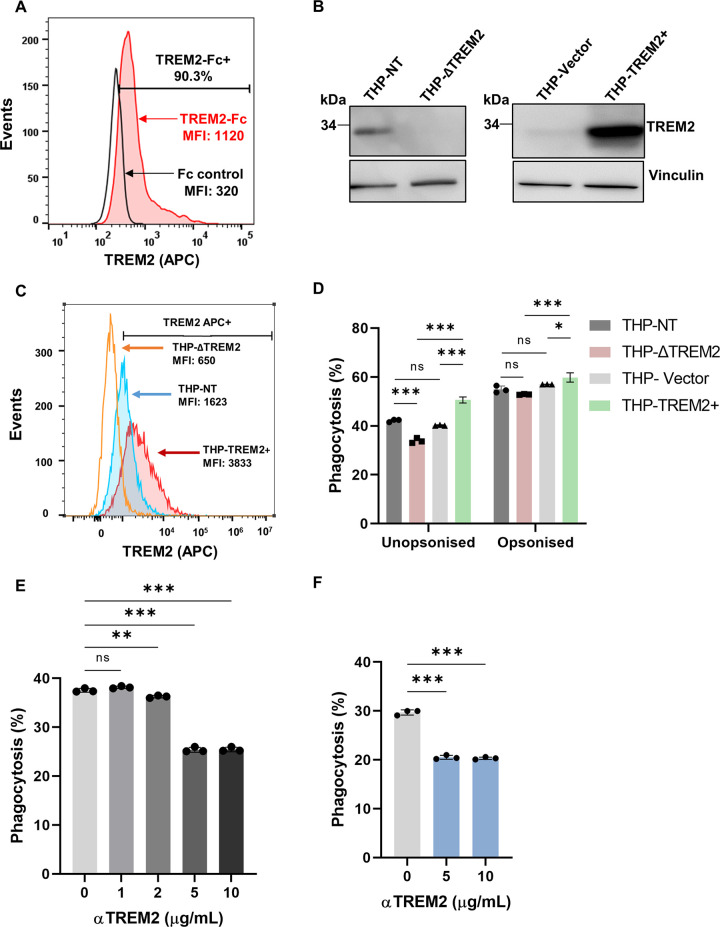
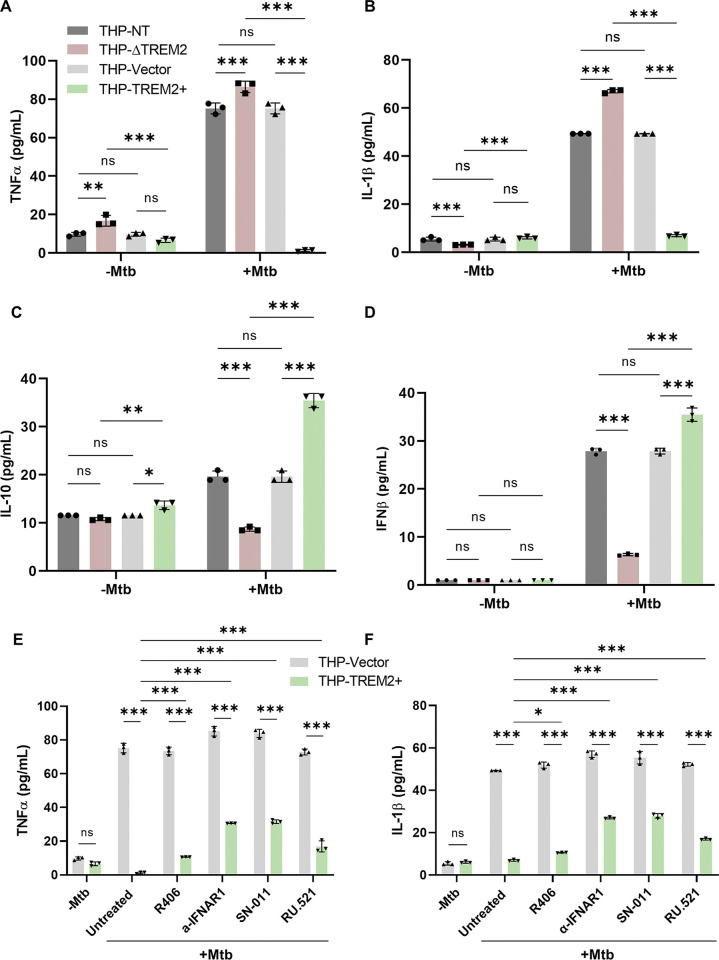
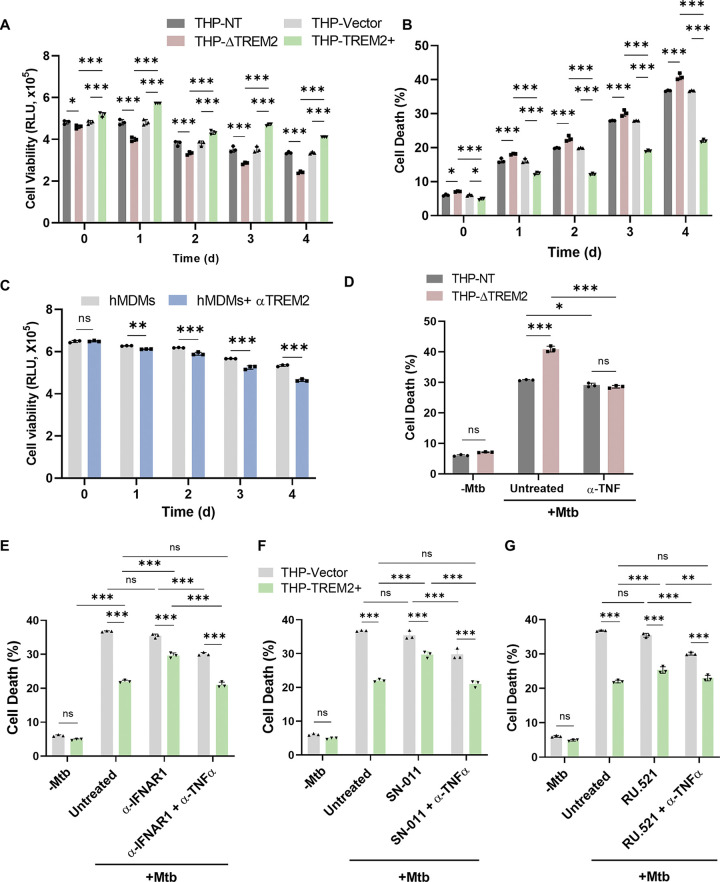
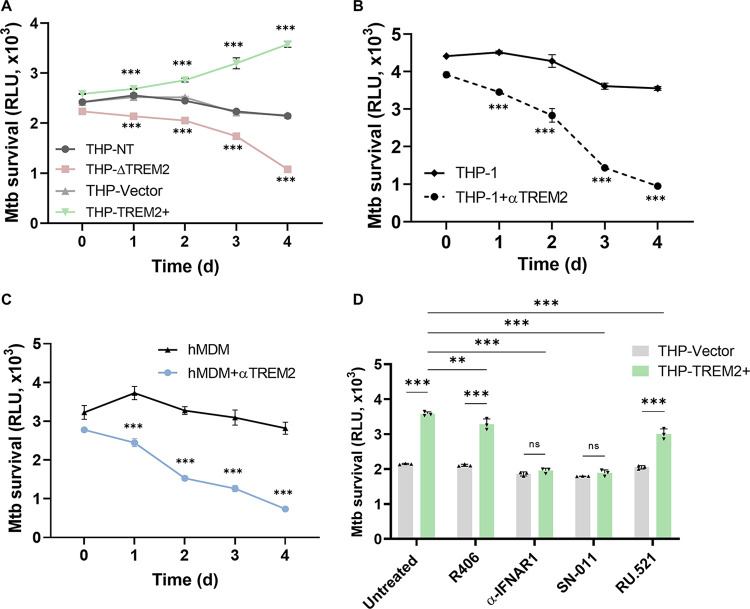
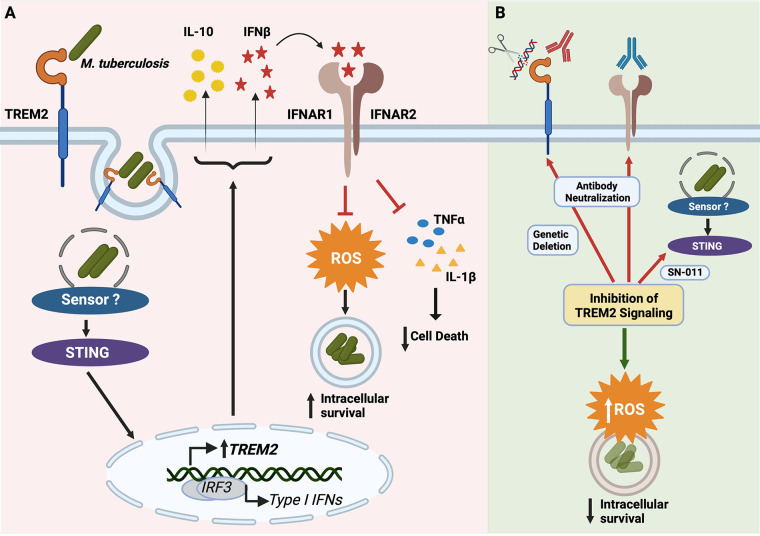
Macrophage surface receptors are critical for pathogen defense, as they are the gatekeepers for pathogen entry and sensing, which trigger robust immune responses. TREM2 (triggering receptor expressed on myeloid cells 2) is a transmembrane surface receptor that mediates anti-inflammatory immune signaling. A recent study showed that TREM2 is a receptor for mycolic acids in the mycobacterial cell wall and inhibits macrophage activation. However, the underlying functional mechanism of how TREM2 regulates the macrophage antimycobacterial response remains unclear. Here, we show that Mycobacterium tuberculosis, the causative agent for tuberculosis, specifically binds to human TREM2 to disable the macrophage antibacterial response. Live but not killed mycobacteria specifically trigger the upregulation of TREM2 during macrophage infection through a mechanism dependent on STING (the stimulator of interferon genes). TREM2 facilitated uptake of M. tuberculosis into macrophages and is responsible for blocking the production of tumor necrosis factor alpha (TNF-α), interleukin-1β (IL-1β), and reactive oxygen species (ROS), while enhancing the production of interferon-β (IFN-β) and IL-10. TREM2-mediated blockade of ROS production promoted the survival of M. tuberculosis within infected macrophages. Consistent with this, genetic deletion or antibody-mediated neutralization of TREM2 reduced the intracellular survival of M. tuberculosis through enhanced production of ROS. Importantly, inhibition of type I IFN signaling in TREM2-overexpressing macrophages restored the ability of these cells to produce inflammatory cytokines and ROS, resulting in normal levels of intracellular bacteria killing. Collectively, our study identifies TREM2 as an attractive host receptor for host-directed antimycobacterial therapeutics. IMPORTANCE Mycobacterium tuberculosis is one of the most ancient bacterial pathogens and remains the leading cause of death from a single bacterial agent. The success of M. tuberculosis relies greatly on its ability to parasitize and disable its host macrophages. Previous studies have found that M. tuberculosis uses its unique cell wall lipids to manipulate the immune response by binding to specific surface receptors on macrophages. Our study reveals that M. tuberculosis binds to TREM2, an immunomodulatory receptor expressed on macrophages, to facilitate a "silent" mode of entry. Increased levels of TREM2 triggered by intracellular sensing of M. tuberculosis promoted the intracellular survival of M. tuberculosis through type I IFN-driven inhibition of reactive oxygen species (ROS) and proinflammatory cytokine production. Importantly, deletion of TREM2 reversed the effects of "silent" entry and resulted in increased production of inflammatory cytokines, generation of ROS, and cell death. As such, antibody-mediated or pharmacological targeting of TREM2 could be a promising strategy for novel treatments against M. tuberculosis infection.
Keywords: IFN; Mycobacterium tuberculosis; TREM2; macrophage cell death; phagocytosis; reactive oxygen species; triggering receptor expressed on myeloid cells; type I interferon.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


Similar articles
-
TREM2 is a receptor for non-glycosylated mycolic acids of mycobacteria that limits anti-mycobacterial macrophage activation.Nat Commun. 2021 Apr 16;12(1):2299. doi: 10.1038/s41467-021-22620-3. Nat Commun. 2021. PMID: 33863908 Free PMC article.
-
CD157 Confers Host Resistance to Mycobacterium tuberculosis via TLR2-CD157-PKCzeta-Induced Reactive Oxygen Species Production.mBio. 2019 Aug 27;10(4):e01949-19. doi: 10.1128/mBio.01949-19. mBio. 2019. PMID: 31455656 Free PMC article.
-
The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis.PLoS Pathog. 2010 Apr 22;6(4):e1000864. doi: 10.1371/journal.ppat.1000864. PLoS Pathog. 2010. PMID: 20421951 Free PMC article.
-
Macrophage takeover and the host-bacilli interplay during tuberculosis.Future Microbiol. 2015;10(5):853-72. doi: 10.2217/fmb.15.11. Future Microbiol. 2015. PMID: 26000654 Review.
-
Role of cytokines in tuberculosis.Immunobiology. 1993 Nov;189(3-4):316-39. doi: 10.1016/S0171-2985(11)80364-5. Immunobiology. 1993. PMID: 8125515 Review.
Cited by
-
Association Between Diabetes Mellitus-Tuberculosis and the Generation of Drug Resistance.Microorganisms. 2024 Dec 20;12(12):2649. doi: 10.3390/microorganisms12122649. Microorganisms. 2024. PMID: 39770852 Free PMC article. Review.
-
Immunological hyporesponsiveness in tuberculosis: The role of mycobacterial glycolipids.Front Immunol. 2022 Dec 2;13:1035122. doi: 10.3389/fimmu.2022.1035122. eCollection 2022. Front Immunol. 2022. PMID: 36544778 Free PMC article. Review.
-
From pathogenesis to antigens: the key to shaping the future of TB vaccines.Front Immunol. 2024 Jul 23;15:1440935. doi: 10.3389/fimmu.2024.1440935. eCollection 2024. Front Immunol. 2024. PMID: 39108269 Free PMC article. Review.
-
Triggering Receptor Expressed on Myeloid Cells 2 Mediates the Involvement of M2-Type Macrophages in Pulmonary Tuberculosis Infection.J Inflamm Res. 2024 Mar 27;17:1919-1928. doi: 10.2147/JIR.S435216. eCollection 2024. J Inflamm Res. 2024. PMID: 38562656 Free PMC article.
-
Endoplasmic Reticulum Stress in Tuberculosis: Molecular Bases and Pathophysiological Implications in the Immunopathogenesis of the Disease.Int J Mol Sci. 2025 May 9;26(10):4522. doi: 10.3390/ijms26104522. Int J Mol Sci. 2025. PMID: 40429667 Free PMC article. Review.
References
-
- WHO. 2020. Global tuberculosis report 2020. https://www.who.int/publications/i/item/9789240013131.
-
- Fabri M, Stenger S, Shin DM, Yuk JM, Liu PT, Realegeno S, Lee H-M, Krutzik SR, Schenk M, Sieling PA, Teles R, Montoya D, Iyer SS, Bruns H, Lewinsohn DM, Hollis BW, Hewison M, Adams JS, Steinmeyer A, Zügel U, Cheng G, Jo E-K, Bloom BR, Modlin RL. 2011. Vitamin D is required for IFN-gamma-mediated antimicrobial activity of human macrophages. Sci Transl Med 3:104ra2. doi:10.1126/scitranslmed.3003045. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
