

De novo variants cause complex symptoms in HSP-ATL1 (SPG3A) and uncover genotype-phenotype correlations
- PMID: 35925862
- PMCID: PMC9838092
- DOI: 10.1093/hmg/ddac182
De novo variants cause complex symptoms in HSP-ATL1 (SPG3A) and uncover genotype-phenotype correlations
Erratum in
-
Erratum to: De novo variants cause complex symptoms in HSP-ATL1 (SPG3A) and uncover genotype-phenotype correlations.Hum Mol Genet. 2023 Sep 5;32(18):2856. doi: 10.1093/hmg/ddad120. Hum Mol Genet. 2023. PMID: 37535873 Free PMC article. No abstract available.
Abstract
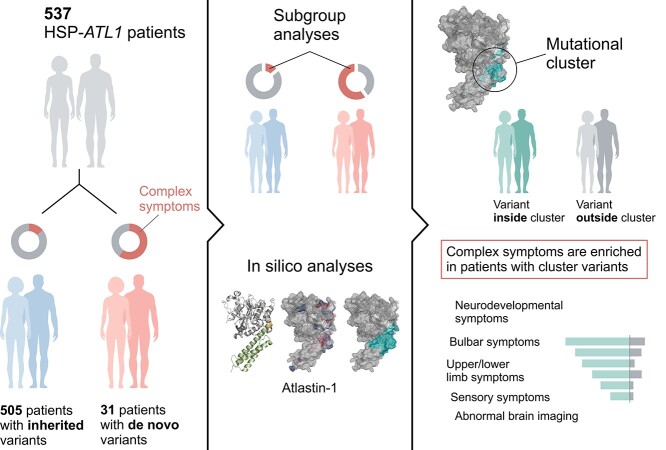
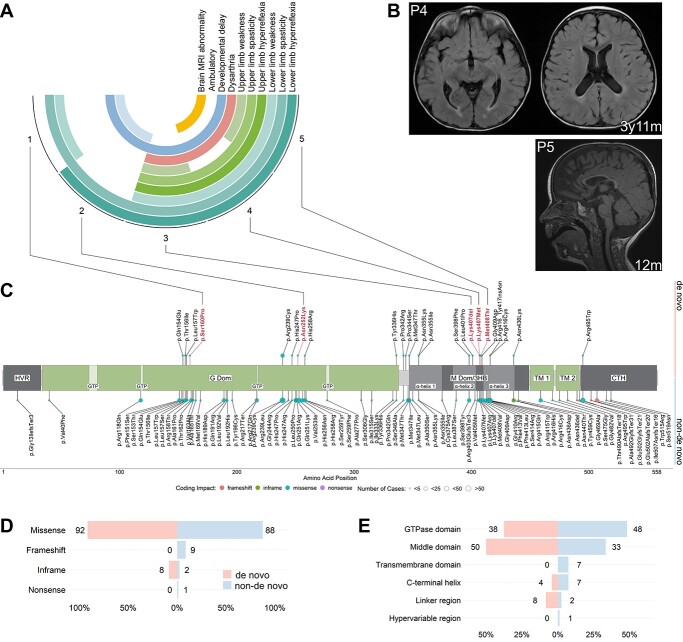
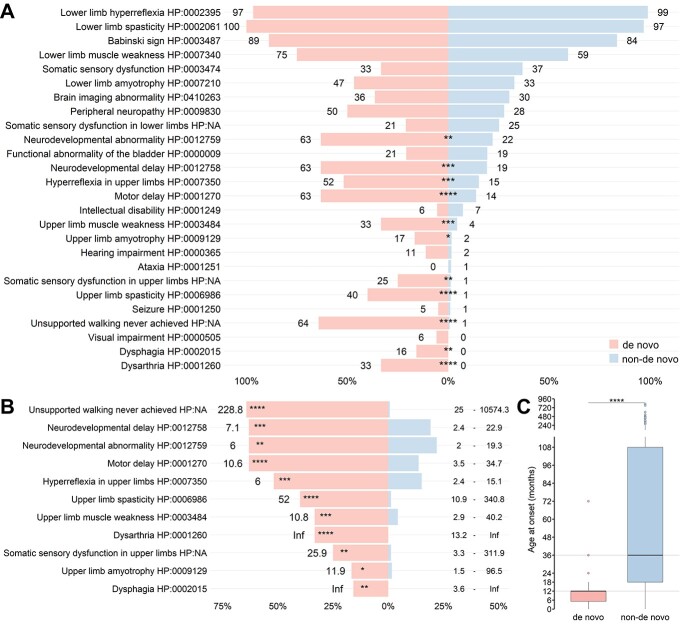
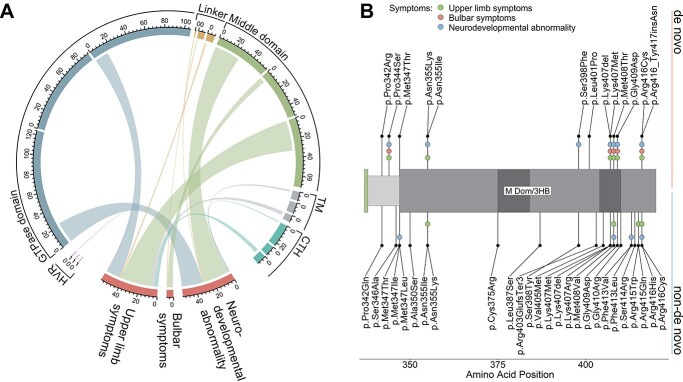
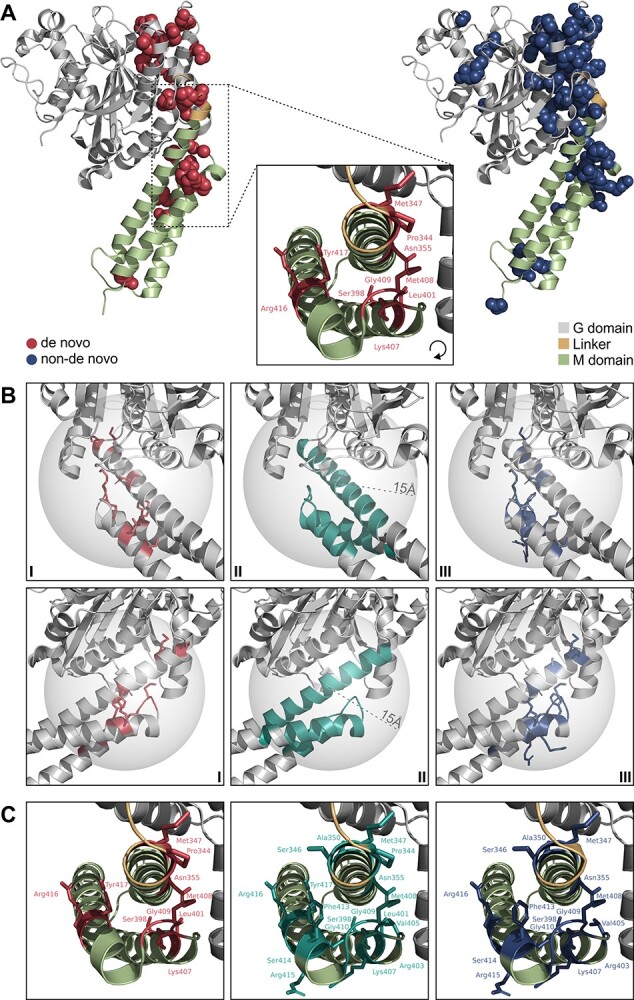
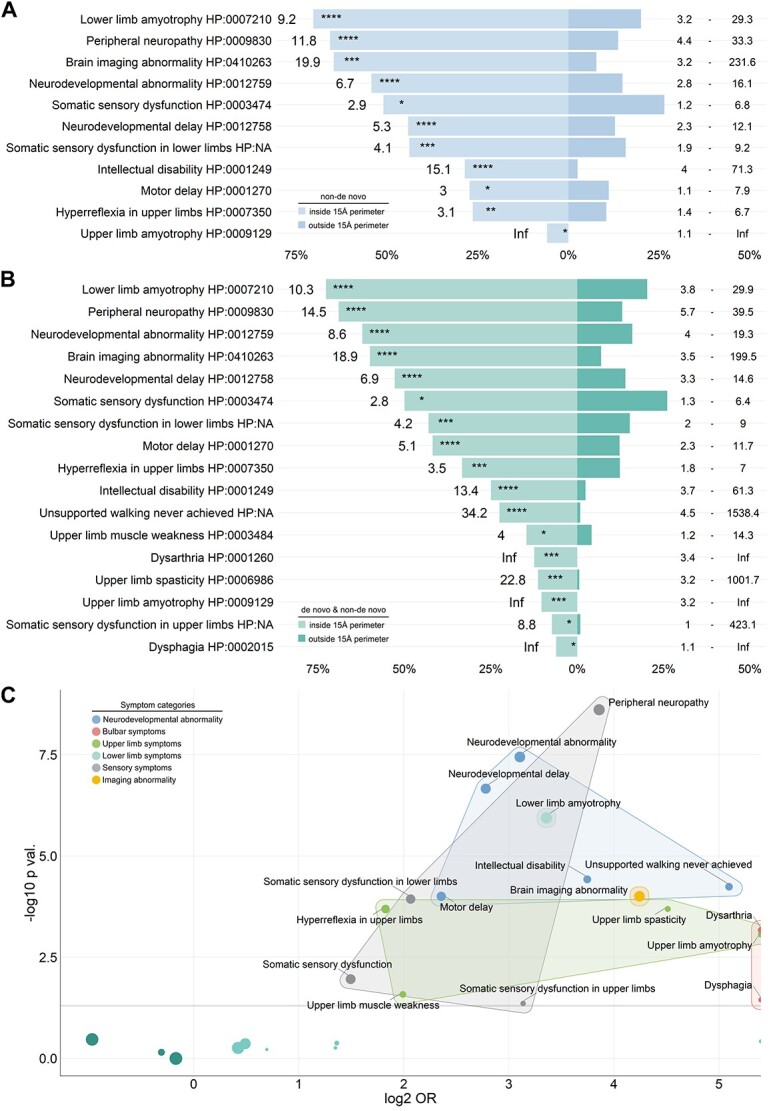
Pathogenic variants in ATL1 are a known cause of autosomal-dominantly inherited hereditary spastic paraplegia (HSP-ATL1, SPG3A) with a predominantly 'pure' HSP phenotype. Although a relatively large number of patients have been reported, no genotype-phenotype correlations have been established for specific ATL1 variants. Confronted with five children carrying de novo ATL1 variants showing early, complex and severe symptoms, we systematically investigated the molecular and phenotypic spectrum of HSP-ATL1. Through a cross-sectional analysis of 537 published and novel cases, we delineate a distinct phenotype observed in patients with de novo variants. Guided by this systematic phenotyping approach and structural modelling of disease-associated variants in atlastin-1, we demonstrate that this distinct phenotypic signature is also prevalent in a subgroup of patients with inherited ATL1 variants and is largely explained by variant localization within a three-dimensional mutational cluster. Establishing genotype-phenotype correlations, we find that symptoms that extend well beyond the typical pure HSP phenotype (i.e. neurodevelopmental abnormalities, upper limb spasticity, bulbar symptoms, peripheral neuropathy and brain imaging abnormalities) are prevalent in patients with variants located within this mutational cluster.
© The Author(s) 2022. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Lange, L.M., Gonzalez-Latapi, P., Rajalingam, R., Tijssen, M.A.J., Ebrahimi-Fakhari, D., Gabbert, C., Ganos, C., Ghosh, R., Kumar, K.R., Lang, A.E. et al. (2022) Nomenclature of genetic movement disorders: recommendations of the International Parkinson and Movement Disorder Society Task Force—an update. Mov. Disord. First published on April 28, 2022, 37, 905–935. - PubMed
-
- Shribman, S., Reid, E., Crosby, A.H., Houlden, H. and Warner, T.T. (2019) Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol., 18, 1136–1146. - PubMed
-
- Erfanian Omidvar, M., Torkamandi, S., Rezaei, S., Alipoor, B., Omrani, M.D., Darvish, H. and Ghaedi, H. (2021) Genotype-phenotype associations in hereditary spastic paraplegia: a systematic review and meta-analysis on 13,570 patients. J. Neurol. First published on November 19, 2019, 268, 2065–2082. - PubMed
-
- Namekawa, M., Ribai, P., Nelson, I., Forlani, S., Fellmann, F., Goizet, C., Depienne, C., Stevanin, G., Ruberg, M., Dürr, A. et al. (2006) SPG3A is the most frequent cause of hereditary spastic paraplegia with onset before age 10 years. Neurology, 66, 112–114. - PubMed
