

Practical Considerations for Single-Cell Genomics
- PMID: 35926125
- PMCID: PMC9479272
- DOI: 10.1002/cpz1.498
Practical Considerations for Single-Cell Genomics
Abstract
The single-cell revolution in the field of genomics is in full bloom, with clever new molecular biology tricks appearing regularly that allow researchers to explore new modalities or scale up their projects to millions of cells and beyond. Techniques abound to measure RNA expression, DNA alterations, protein abundance, chromatin accessibility, and more, all with single-cell resolution and often in combination. Despite such a rapidly changing technology landscape, there are several fundamental principles that are applicable to the majority of experimental workflows to help users avoid pitfalls and exploit the advantages of the chosen platform. In this overview article, we describe a variety of popular single-cell genomics technologies and address some common questions pertaining to study design, sample preparation, quality control, and sequencing strategy. As the majority of relevant publications currently revolve around single-cell RNA-seq, we will prioritize this genomics modality in our discussion. © 2022 Wiley Periodicals LLC.
Keywords: genomics; scATAC-seq; scRNA-seq; sequencing; single-cell; transcriptomics.
© 2022 Wiley Periodicals LLC.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT:
The authors declare no conflicts of interest.
Figures
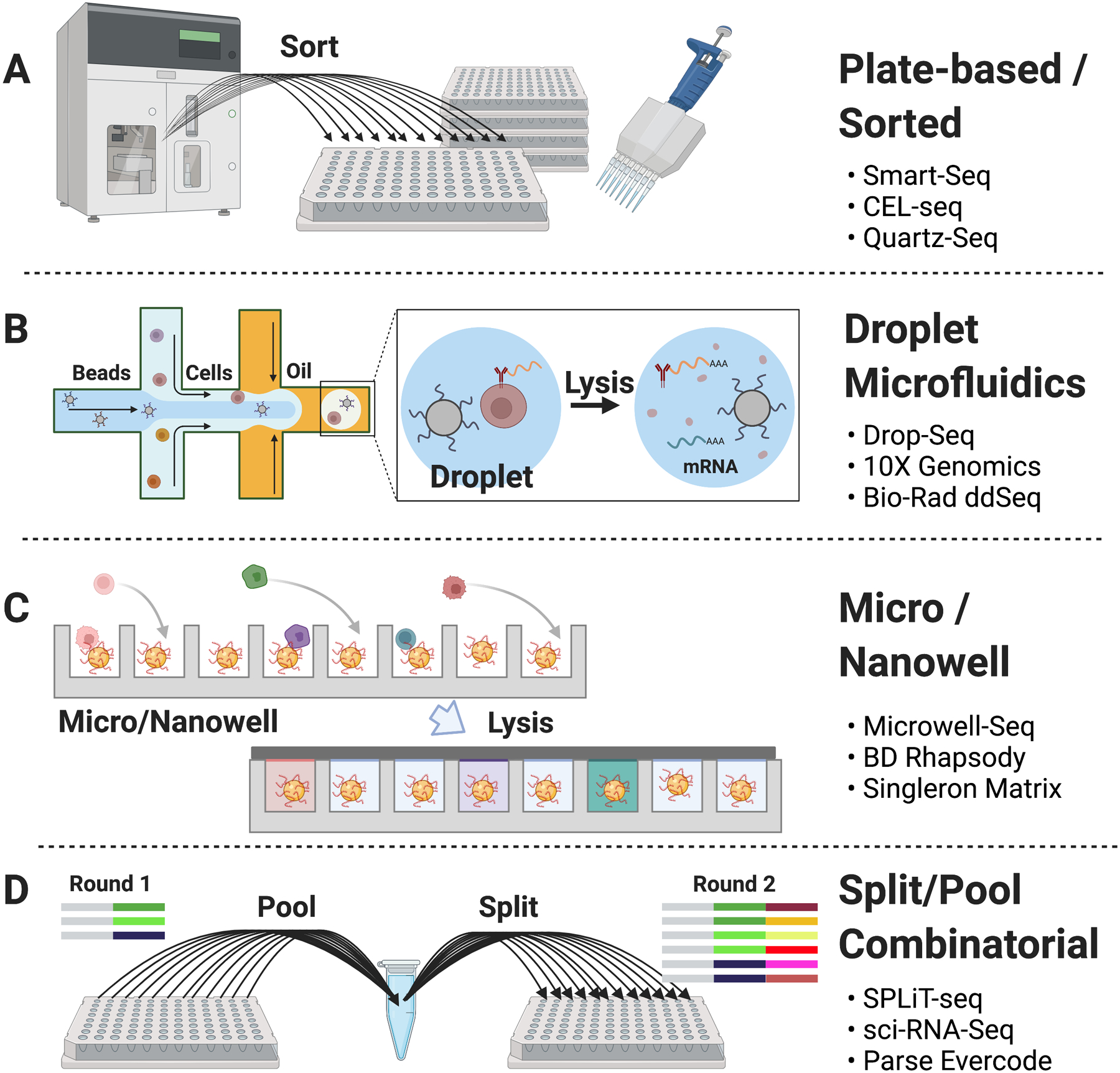
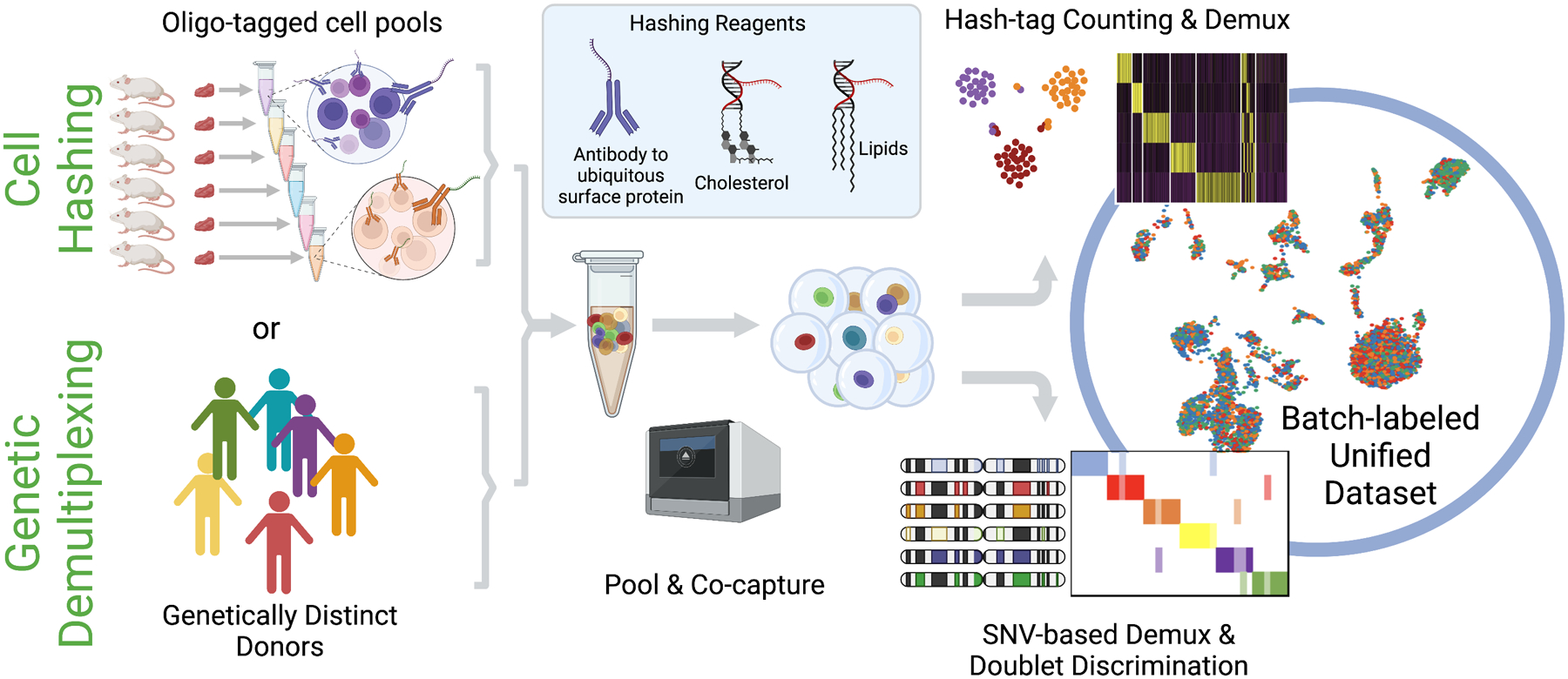
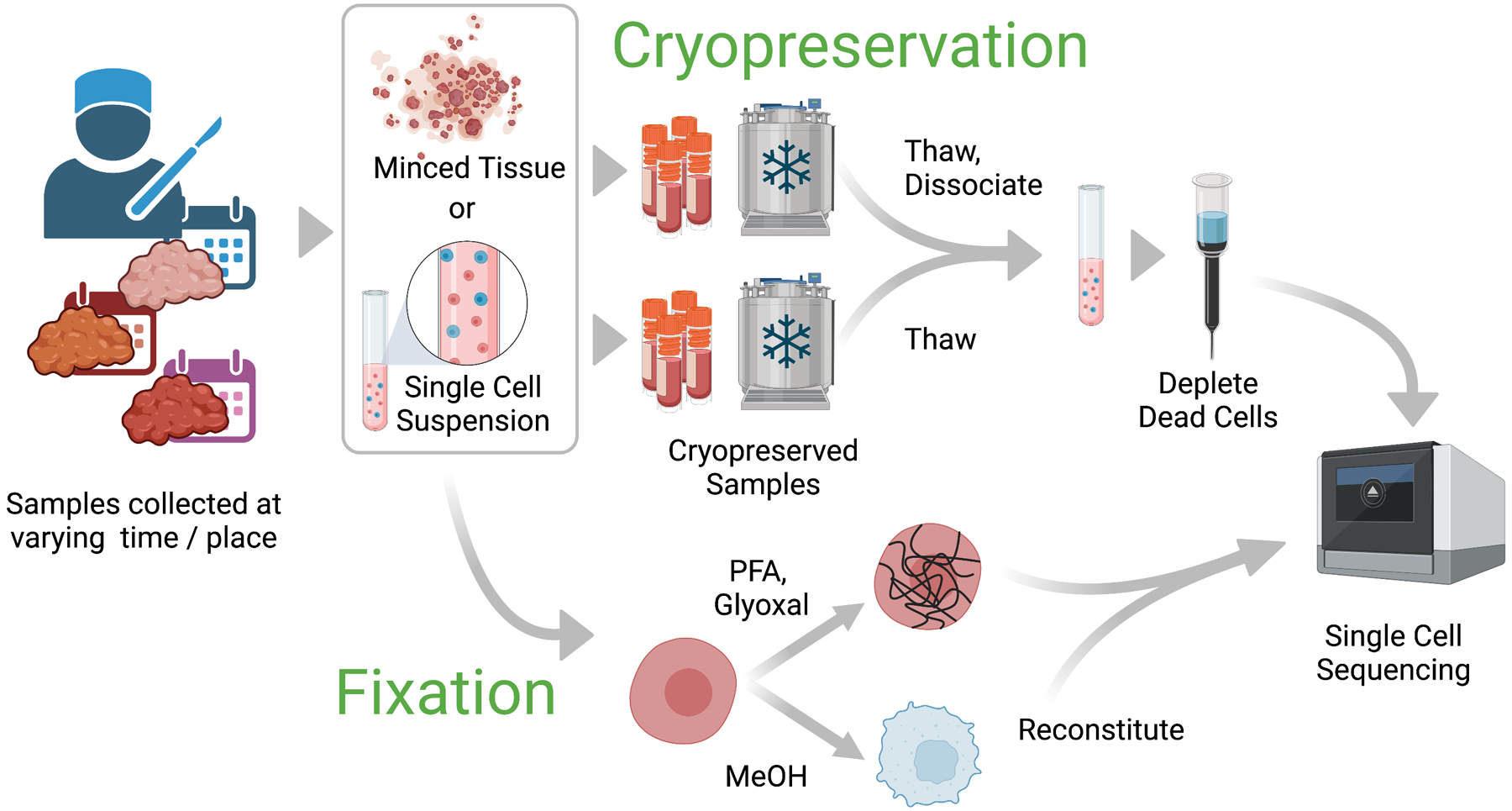
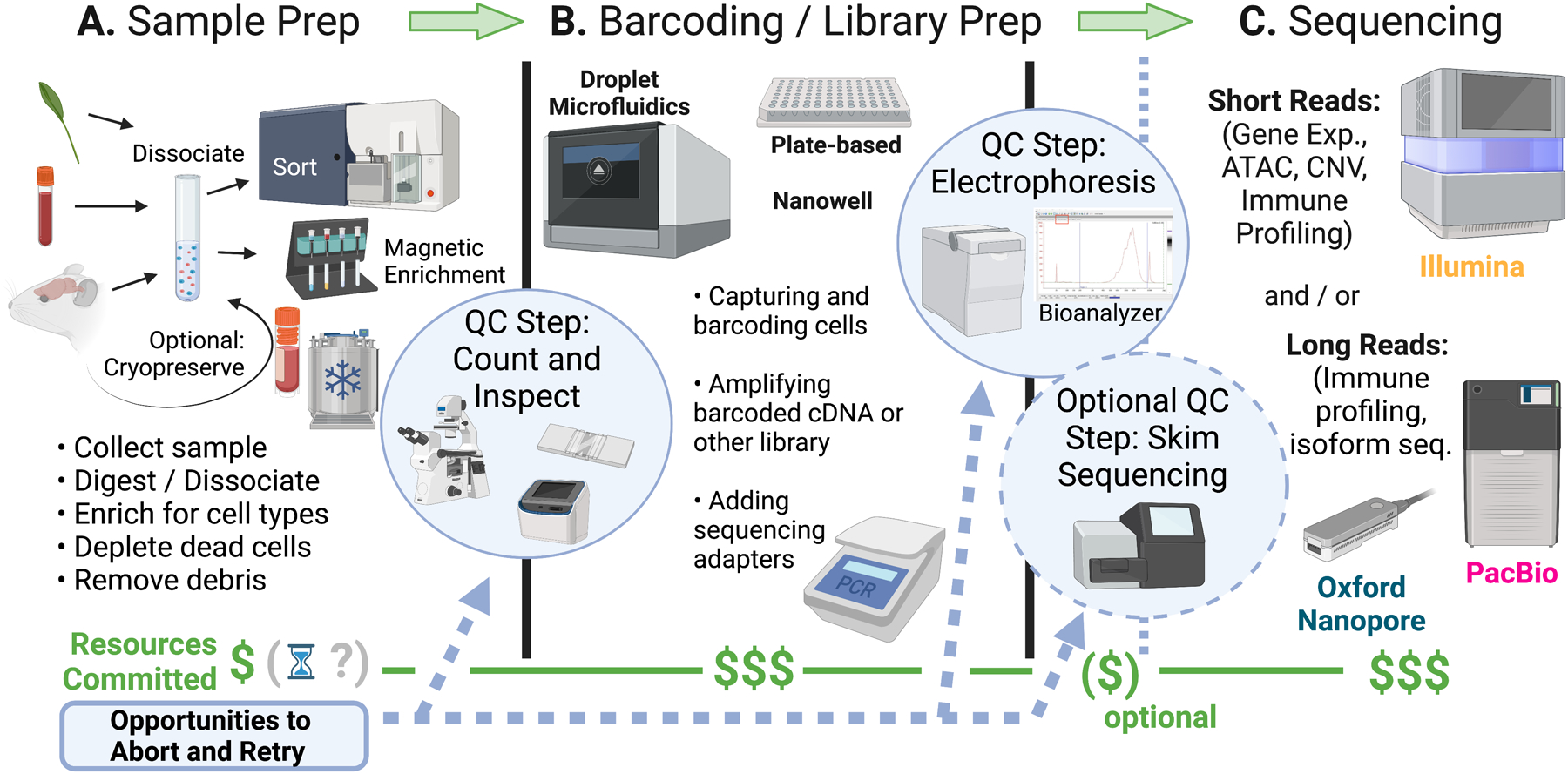
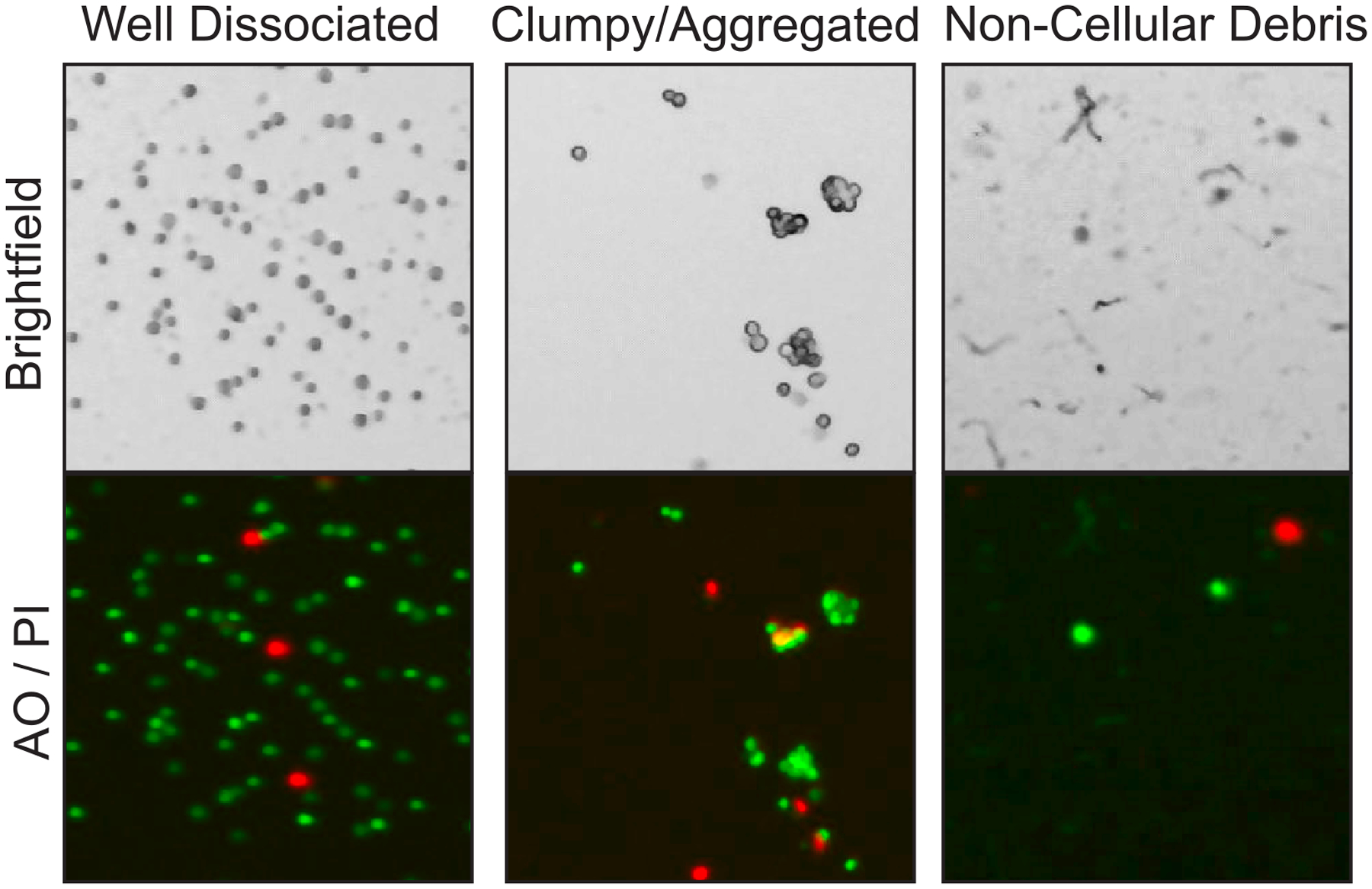


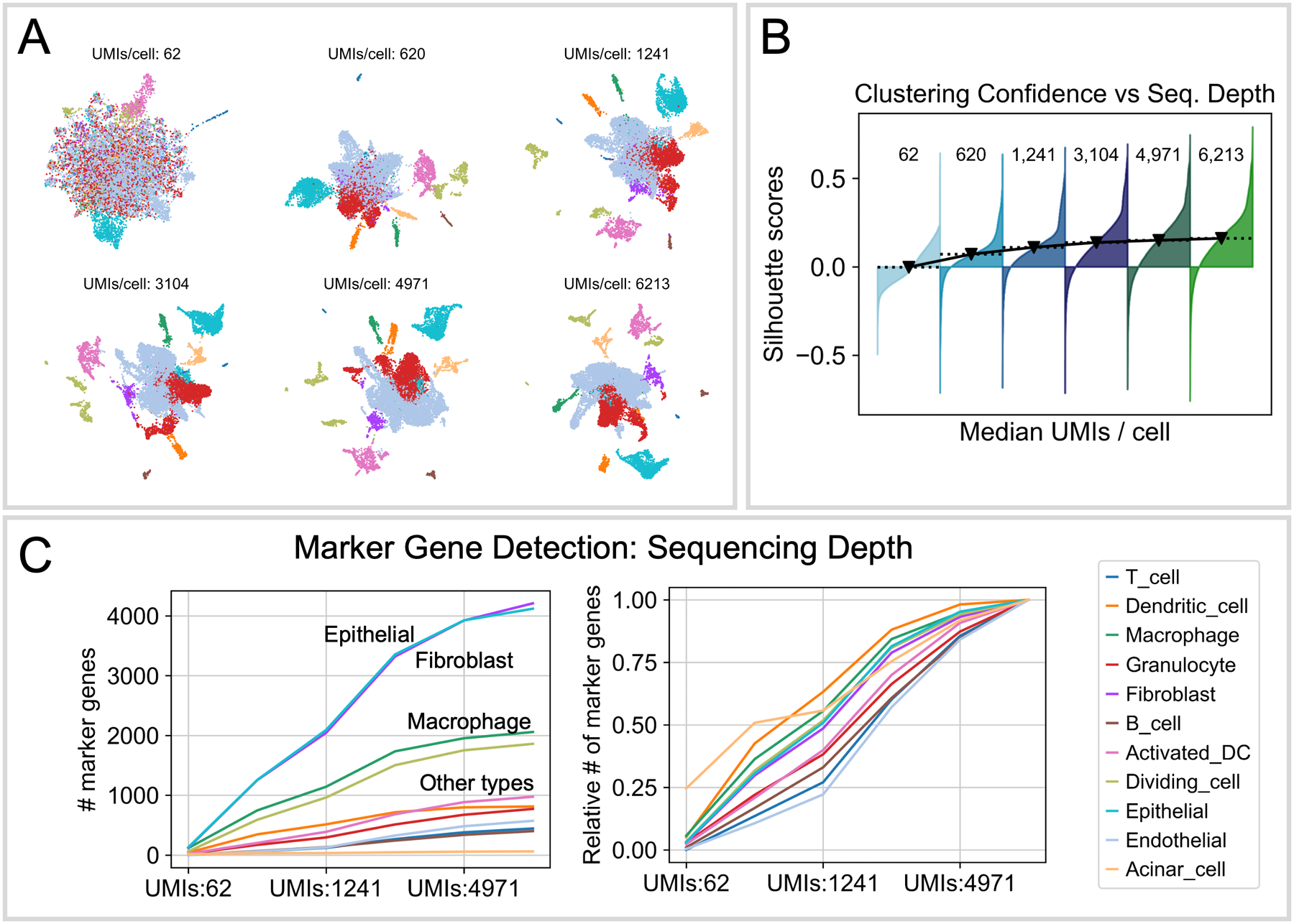
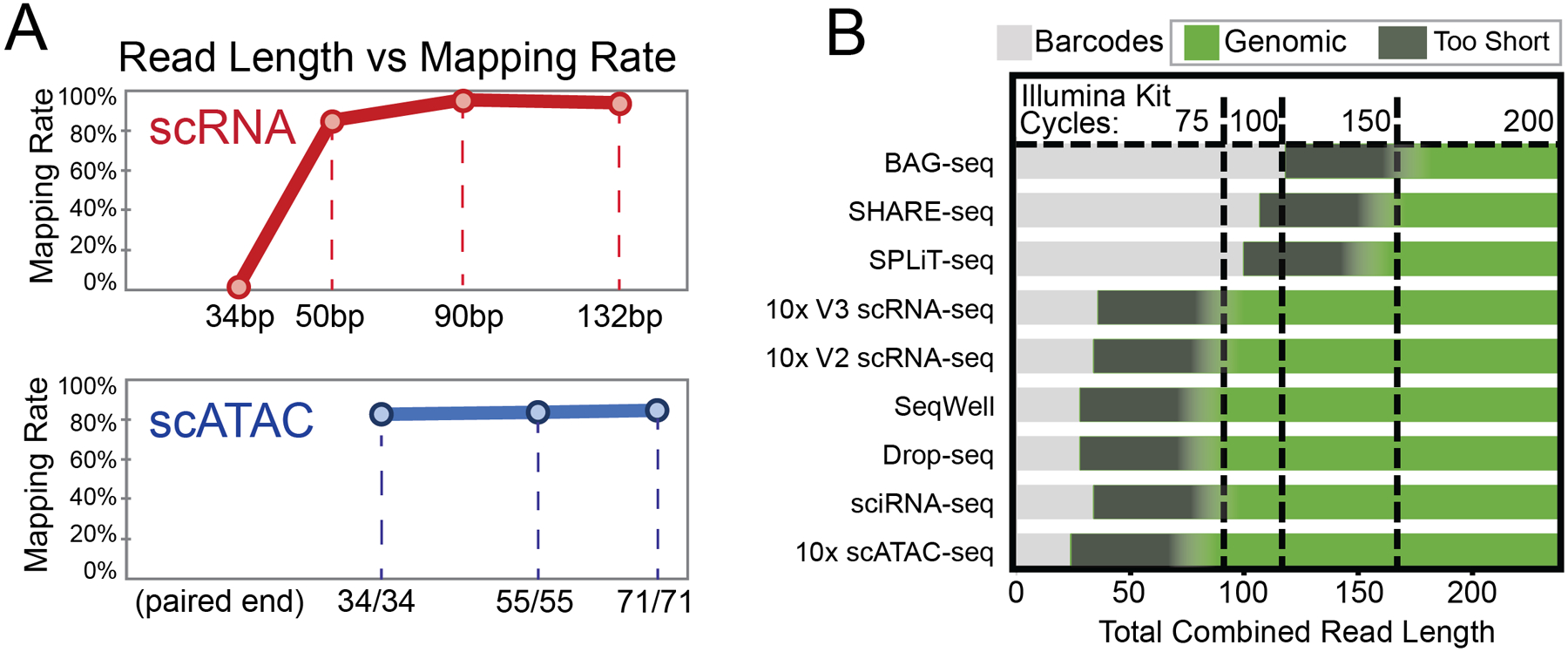
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
