

Cancer genes disfavoring T cell immunity identified via integrated systems approach
- PMID: 35926468
- PMCID: PMC9402397
- DOI: 10.1016/j.celrep.2022.111153
Cancer genes disfavoring T cell immunity identified via integrated systems approach
Abstract
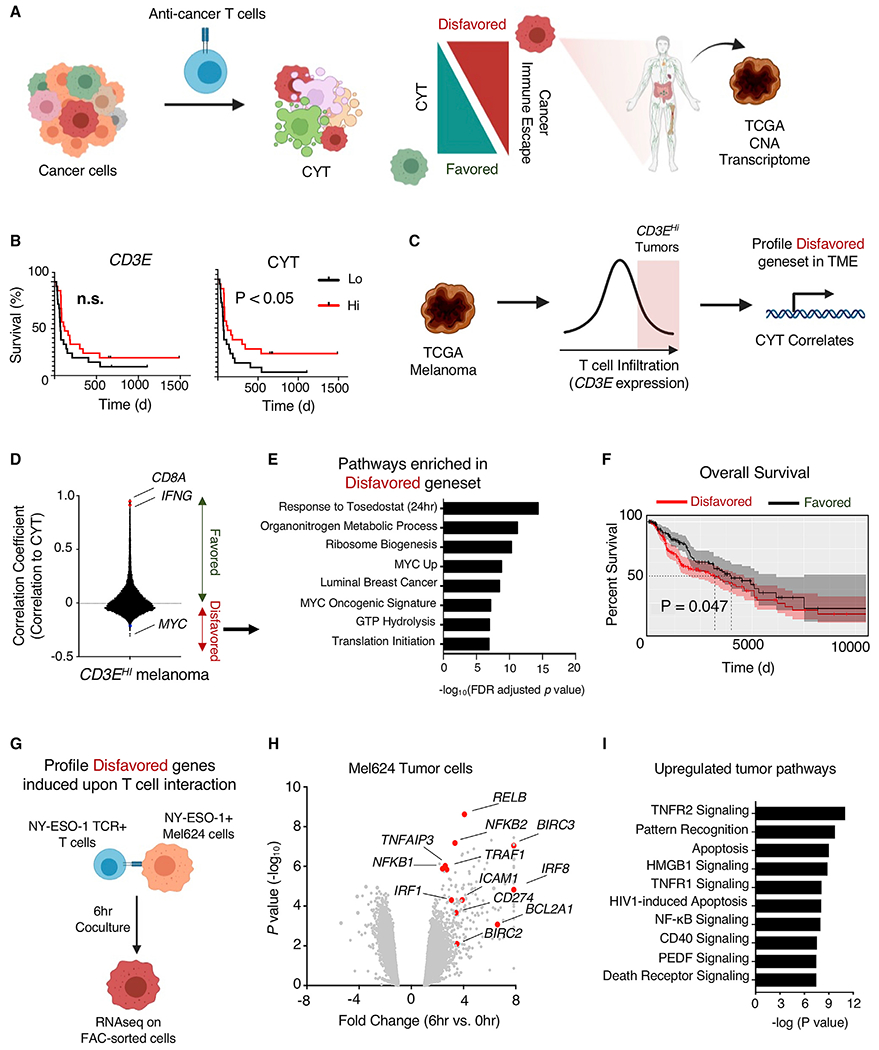
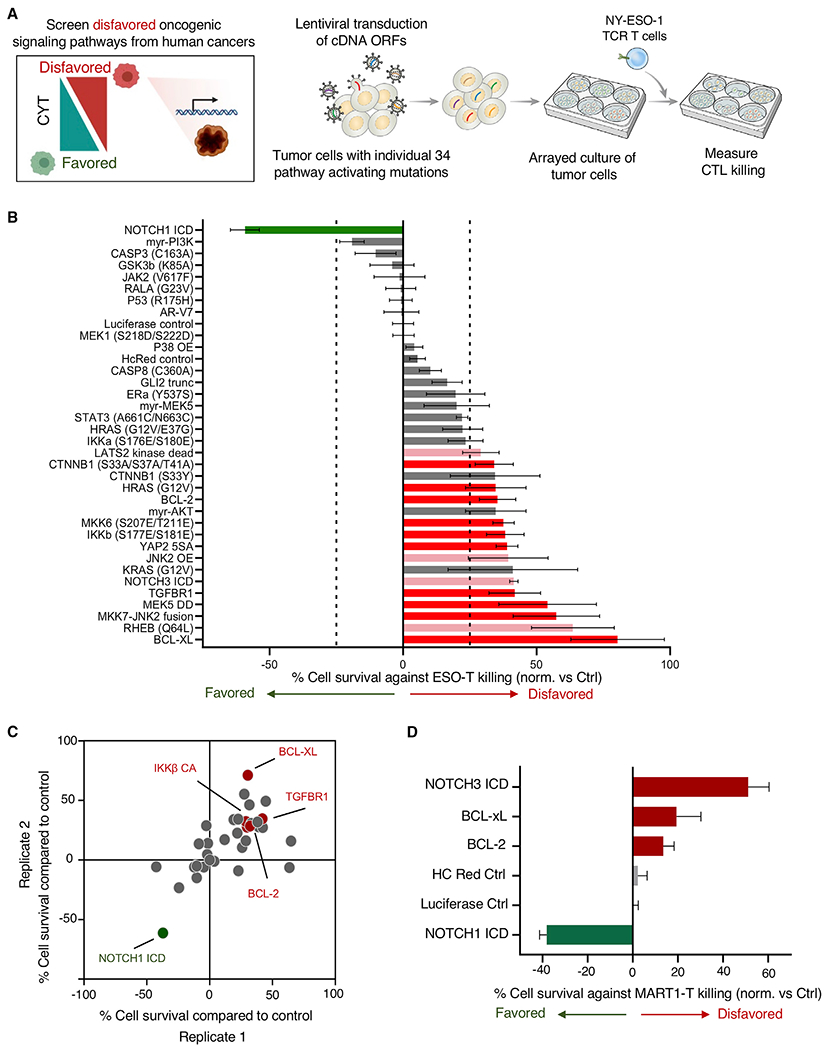
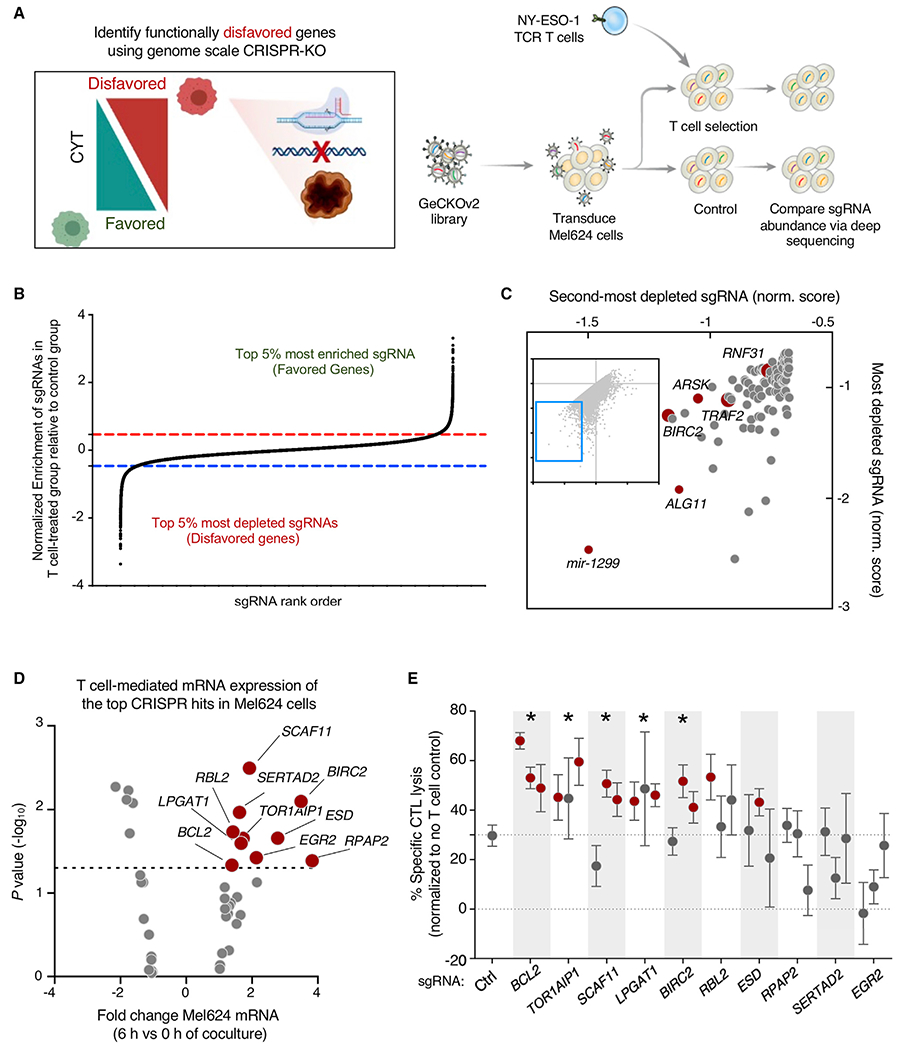
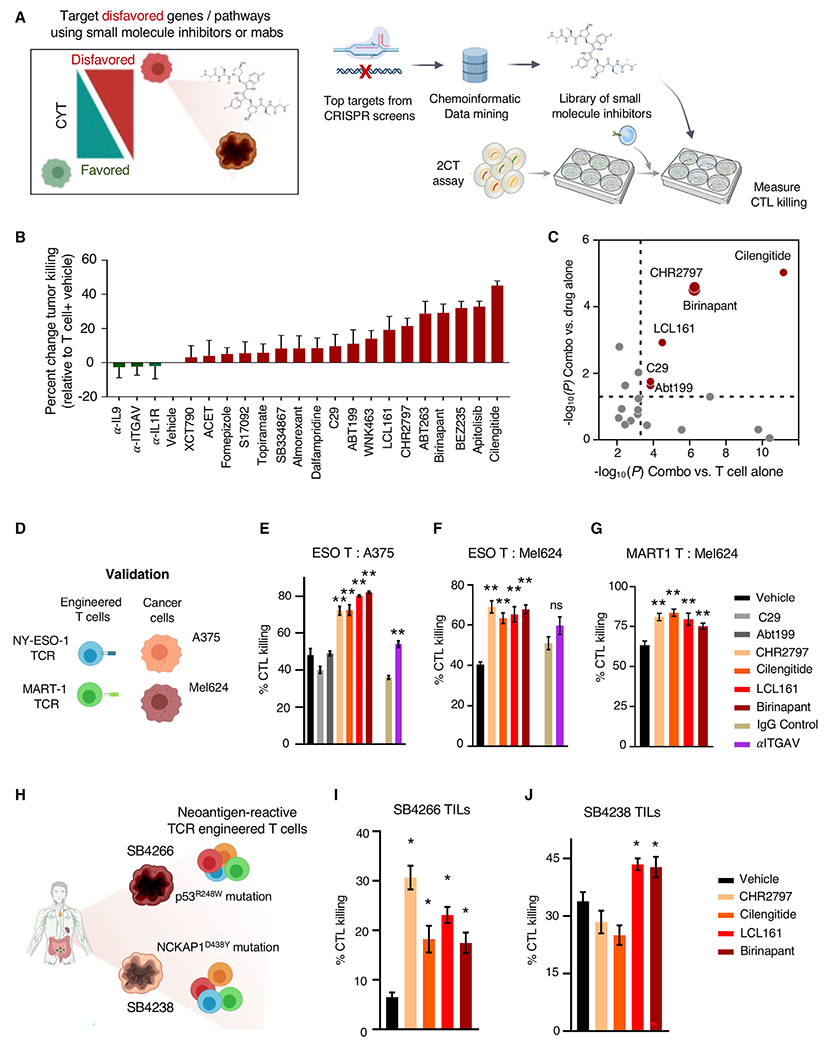
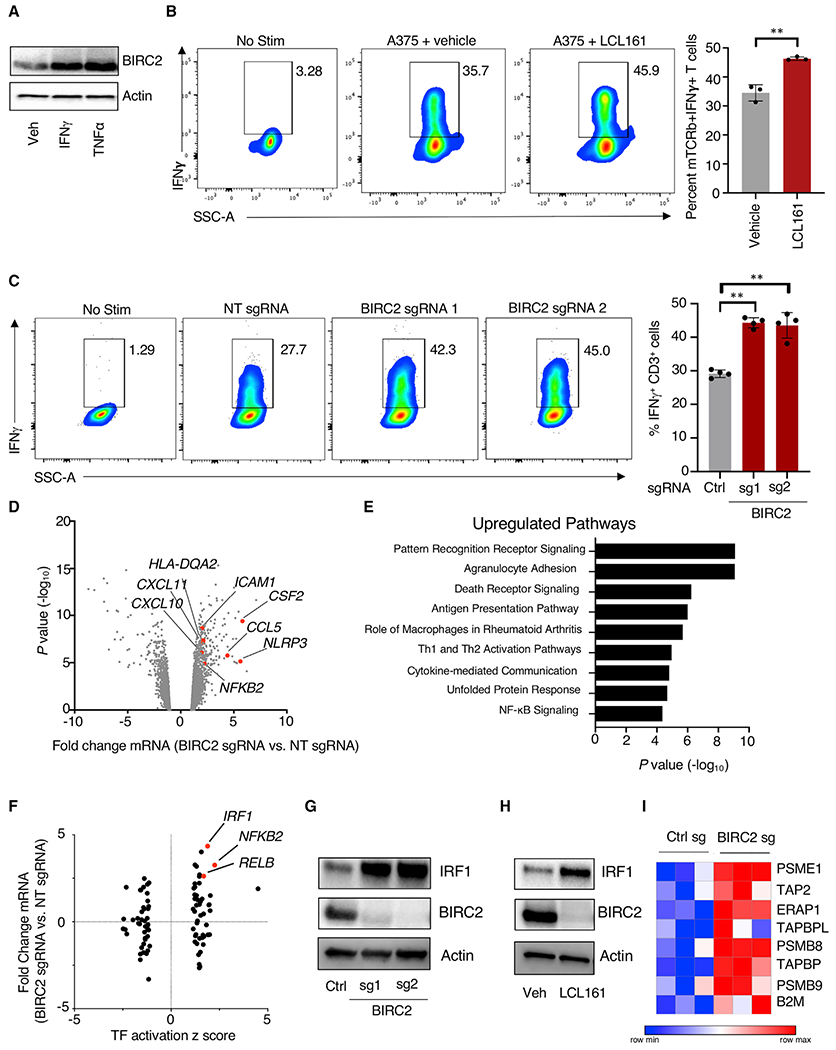
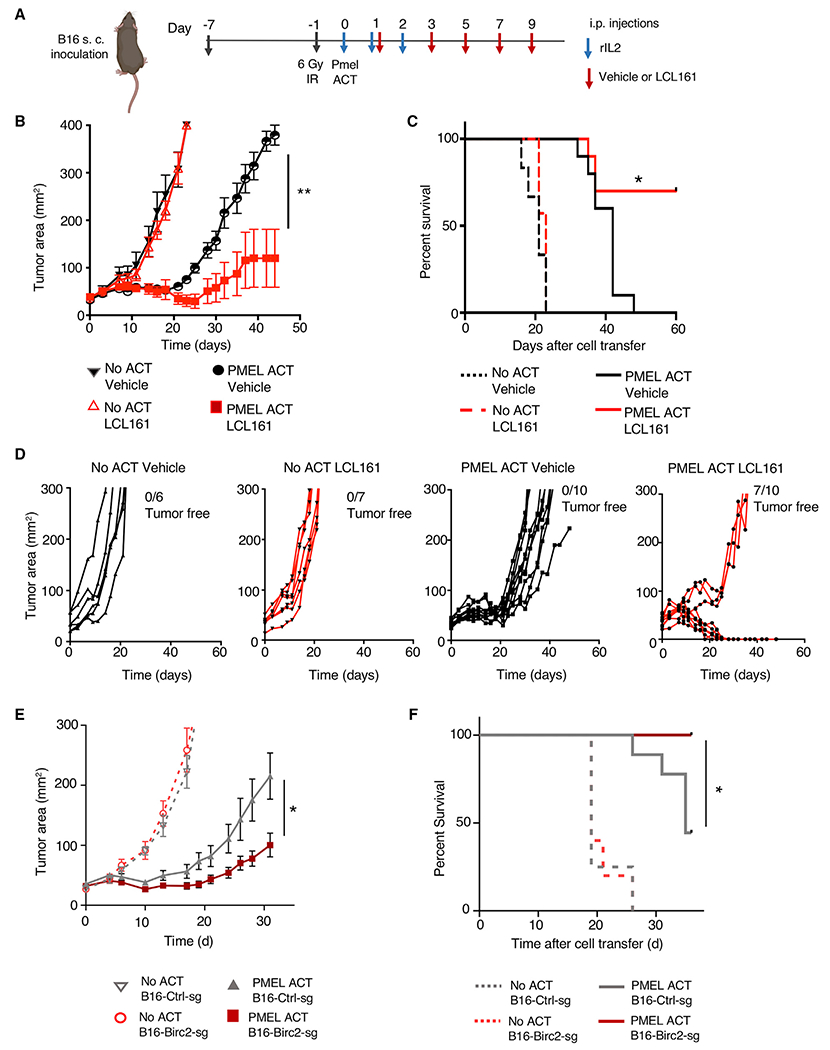
Adoptive T cell therapies (ACT) have been curative for a limited number of cancer patients. The sensitization of cancer cells to T cell killing may expand the benefit of these therapies for more patients. To this end, we use a three-step approach to identify cancer genes that disfavor T cell immunity. First, we profile gene transcripts upregulated by cancer under selection pressure from T cell killing. Second, we identify potential tumor gene targets and pathways that disfavor T cell killing using signaling pathway activation libraries and genome-wide loss-of-function CRISPR-Cas9 screens. Finally, we implement pharmacological perturbation screens to validate these targets and identify BIRC2, ITGAV, DNPEP, BCL2, and ERRα as potential ACT-drug combination candidates. Here, we establish that BIRC2 limits antigen presentation and T cell recognition of tumor cells by suppressing IRF1 activity and provide evidence that BIRC2 inhibition in combination with ACT is an effective strategy to increase efficacy.
Keywords: CP: Cancer; CP: Immunology; CRISPR screen; cell therapy; combination immunotherapy; gain-of-function screen; immunotherapy resistance.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of interests R.J.K., S.K.V., Y.P., and N.P.R. hold equity in and are currently employed by Lyell Immunopharma, South San Francisco, CA, USA. S.J.P. has previously held positions at Boehringer Ingelheim and NextCure Inc. and currently holds equity in NextCure Inc. N.E.S. is a scientific advisor for Vertex and Qiagen.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
