

Ca2+ homeostasis maintained by TMCO1 underlies corpus callosum development via ERK signaling
- PMID: 35927240
- PMCID: PMC9352667
- DOI: 10.1038/s41419-022-05131-x
Ca2+ homeostasis maintained by TMCO1 underlies corpus callosum development via ERK signaling
Abstract
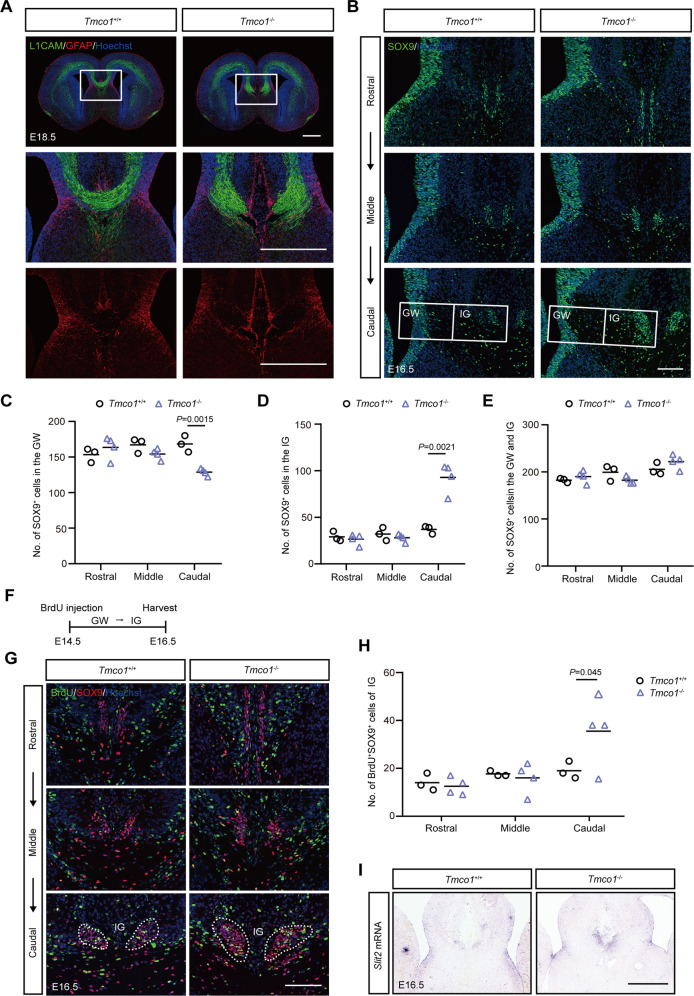
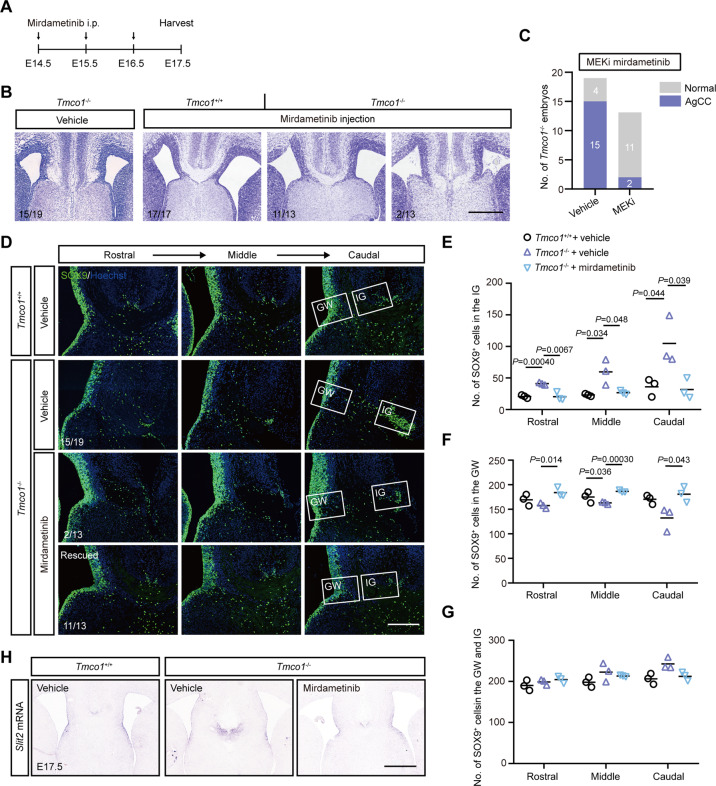
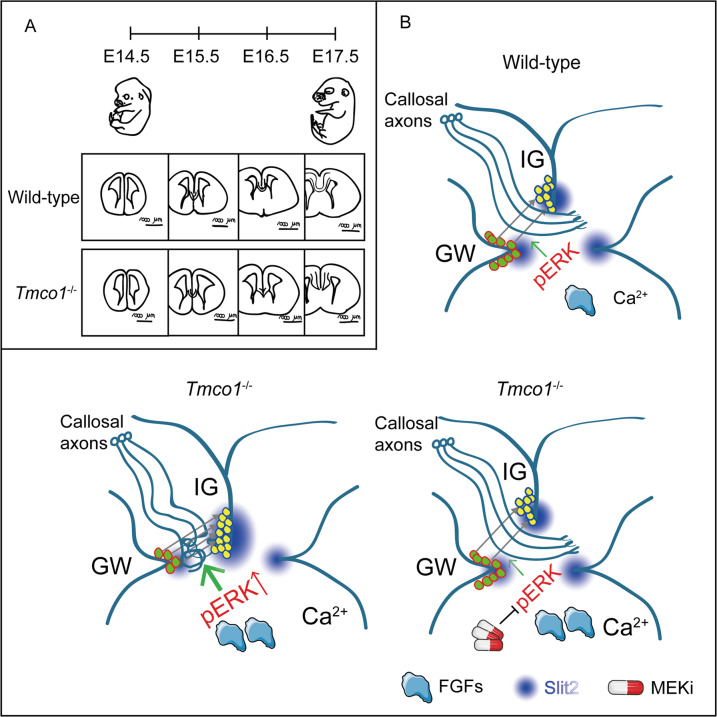
Transmembrane of coiled-coil domains 1 (TMCO1) plays an important role in maintaining homeostasis of calcium (Ca2+) stores in the endoplasmic reticulum (ER). TMCO1-defect syndrome shares multiple features with human cerebro-facio-thoracic (CFT) dysplasia, including abnormal corpus callosum (CC). Here, we report that TMCO1 is required for the normal development of CC through sustaining Ca2+ homeostasis. Tmco1-/- mice exhibit severe agenesis of CC with stalled white matter fiber bundles failing to pass across the midline. Mechanistically, the excessive Ca2+ signals caused by TMCO1 deficiency result in upregulation of FGFs and over-activation of ERK, leading to an excess of glial cell migration and overpopulated midline glia cells in the indusium griseum which secretes Slit2 to repulse extension of the neural fiber bundles before crossing the midline. Supportingly, using the clinical MEK inhibitors to attenuate the over-activated FGF/ERK signaling can significantly improve the CC formation in Tmco1-/- brains. Our findings not only unravel the underlying mechanism of abnormal CC in TMCO1 defect syndrome, but also offer an attractive prevention strategy to relieve the related agenesis of CC in patients.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



References
-
- Gerhard B, Elżbieta P-P, Richard B, Iris U, Giorgi K. Corpus callosum and epilepsies. J Epileptol. 2013;21:89–104. doi: 10.1515/joepi-2015-0008. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
