

TP53-dependent toxicity of CRISPR/Cas9 cuts is differential across genomic loci and can confound genetic screening
- PMID: 35927263
- PMCID: PMC9352712
- DOI: 10.1038/s41467-022-32285-1
TP53-dependent toxicity of CRISPR/Cas9 cuts is differential across genomic loci and can confound genetic screening
Abstract
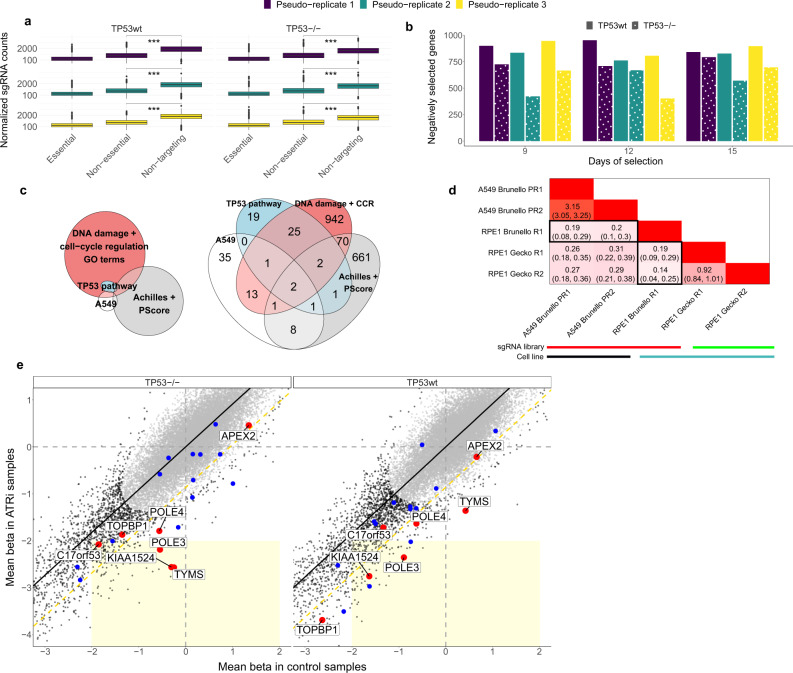
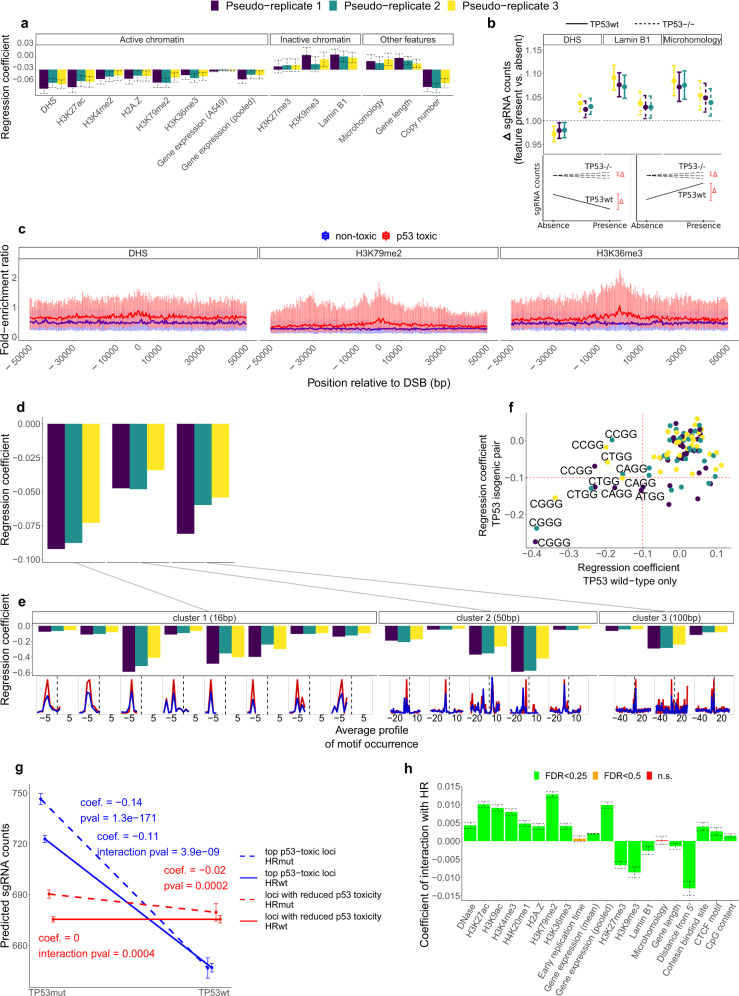
CRISPR/Cas9 gene editing can inactivate genes in a precise manner. This process involves DNA double-strand breaks (DSB), which may incur a loss of cell fitness. We hypothesize that DSB toxicity may be variable depending on the chromatin environment in the targeted locus. Here, by analyzing isogenic cell line pair CRISPR experiments jointly with previous screening data from across ~900 cell lines, we show that TP53-associated break toxicity is higher in genomic regions that harbor active chromatin, such as gene regulatory elements or transcription elongation histone marks. DSB repair pathway choice and DNA sequence context also associate with toxicity. We also show that, due to noise introduced by differential toxicity of sgRNA-targeted sites, the power of genetic screens to detect conditional essentiality is reduced in TP53 wild-type cells. Understanding the determinants of Cas9 cut toxicity will help improve design of CRISPR reagents to avoid incidental selection of TP53-deficient and/or DNA repair deficient cells.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
